
Вирусный лейкоз крупного рогатого скота (ВЛ КРС)
Лейкоз крупного рогатого скота — хроническая вирусная болезнь.
Различают три стадии (периода) в развитии инфекции: инкубационную, гематологическую и опухолевую.
Источником возбудителя болезни являются зараженные вирусом лейкоза крупного рогатого скота животные на всех стадиях инфекционного процесса.
Животные заражаются при проникновении в организм лимфоцитов, содержащих вирус лейкоза.
Болезнь протекает бессимптомно, характеризуется злокачественным разрастанием кроветворных и лимфоидных клеток в разных органах.
В ходе воспалительных процессов ухудшается общее состояние животного, снижается удой, нередко наблюдается истощение. У больных коров снижается уровень иммунитета и, как следствие, увеличивается яловость, снижается выход телят, учащаются аборты.
Контагионность лейкоза крупного рогатого скота незначительная.
Болезнь регистрируется практически во всех странах мира. В России сегодня вирус распространен почти во всех субъектах.Территория Санкт‑Петербурга благополучная по вирусному лейкозу крупного рогатого скота.
В настоящее время лейкоз крупного рогатого скота – большая социально-биологическая проблема в России!
Лейкоз крупного рогатого скота – болезнь, потенциально опасная для человека.
Выявить болезнь на ранних стадиях можно путем проведения диагностических исследований.
Государственной ветеринарной службой проводятся плановые исследования на лейкоз крупного рогатого скота, содержащегося организациями и физическими лицами на территории Санкт‑Петербурга.
Если Вы содержите крупный рогатый скот или знаете, кто из Ваших соседей содержит, — сообщите нам об этом!
Если в Вашем личном подворье заболели животные – сообщите нам об этом!
Государственная ветеринарная служба Санкт‑Петербурга
звоните нам: 717-52-10, 374-98-23, 527-50-45
Лейкоз крупного рогатого скота ликвидировали в 22 хозяйствах Подмосковья в 2020 году
В Московской области оздоровлено от лейкоза крупного рогатого скота 22 хозяйства по итогам 2020 года, сообщает пресс-служба регионального Министерства сельского хозяйства и продовольствия.
«Одной из важнейших программ по безопасности пищевой продукции является оздоровление и ликвидация лейкоза крупного рогатого скота в неблагополучных хозяйствах Московской области. В декабре 2020 года оздоровлено от лейкоза два хозяйства. Это «Руссмолоко» Сергиево-Посадского городского округа и «Красногорский завод имени С.А. Зверева» Волоколамска. Всего по итогам года оздоровлено 22 пункта по лейкозу», — сообщил исполняющий обязанности министра сельского хозяйства и продовольствия Подмосковья Сергей Воскресенский.
Вирус лейкоза крупного рогатого скота — хроническая инфекционная болезнь, которая характеризуется злокачественным разрастанием клеток кроветворных органов.
«Оздоровление хозяйств проводится за счет собственного воспроизводства стада и субсидий из бюджета области на приобретение коров и нетелей для замены инфицированных лейкозом животных. Для определения благополучия поголовья скота руководители племенных и нетелиных комплексов, владельцы, занимающиеся реализацией животных, обязаны обеспечить ежегодное однократное проведение клинических осмотров и исследований всех животных старше шестимесячного возраста», — отметил Воскресенский.
Ветеринарные специалисты региона предлагают эффективные пути решения по оздоровлению крупного рогатого скота и проводят систематическое карантинирование и дезинфекцию животноводческих помещений и ферм.
«На сегодняшний день в Подмосковье 19 хозяйств остаются неблагополучными по лейкозу крупного рогатого скота. За два года оздоровлено 46 неблагополучных хозяйств», — заключил Воскресенский.
Московская область является лидером по темпам оздоровления хозяйств от лейкоза КРС.
Как получить удостоверение тракториста в Подмосковье>>
ЛЕЙКОЗ крупного рогатого скота
Лейкоз (лейкемия, белокровие) млекопитающих – вирусное опухолевое заболевание гемолимфатической системы, характеризующееся злокачественным разрастанием кроветворной ткани, нарушением процессов созревания (не регулируемым ростом) кровяных клеток и выходом в кровь большого числа незрелых форменных элементов крови.
Лейкозом болеют все виды млекопитающих животных, птицы, рыбы, рептилии.
Источником возбудителя лейкоза является больное животное. Уже на ранних стадиях болезни вирус выделяется из организма коров с молозивом и молоком. Имеются сведения о возможном выделении возбудителя со слюной. Внутри стад возможны вертикальный (через плаценту, молозиво и молоко) и горизонтальный механизмы передачи возбудителя, однако конгениальное заражение происходит сравнительно редко. (Термин конгениальное означает «присутствующий при рождении»).
Телята заражаются главным образом через плаценту.
Особенностью лейкоза является длительное течение болезни без заметных нарушений в состоянии здоровья животных.
При клиническом осмотре у больных животных обнаруживают увеличение поверхностных лимфатических узлов (предлопаточных, надколенных, надвымянных, подчелюстных), которое может быть 2х-3х кратным. Лимфоузлы безболезненны, плотной консистенции, подвижны.
Главной проблемой в профилактике и ликвидации лейкоза крупного рогатого скота является значительное инфицирование вирусом крупного рогатого скота, не имеющего признаков болезни.
Диагноз в благополучном по лейкозу хозяйстве устанавливается на основании положительных результатов серологического и гематологического исследований или патоморфологического исследований.
Для поддержания благополучия владельцы животных обязаны обеспечить ежегодное проведение клинических осмотров и серологических исследований всех животных старше 6- месячного возраста. Из благополучных по лейкозу хозяйств животные реализуются без ограничений. При этом за 30 дней до вывоза животных из хозяйства их подвергают серологическому исследованию на лейкоз методом РИД (реакция иммунной диффузии). Завоз животных необходимо осуществлять с ветеринарными сопроводительными документами.
С целью роста и развития молочного животноводства на территории СО Департаментом ветеринарии совместно МинАПК области принято решение о проведении в 2020 году в обязательном порядке двукратных серологических исследований на лейкоз всего восприимчивого поголовья.
Столь распространённое заболевание как лейкоз у коров, до сих пор до конца не изучено. До некоторого времени считалось, что лейкоз коров не опасен для человека. Последние исследования показали, что есть опасность заражения этим заболеванием для людей. Поэтому приобретает значение своевременная диагностика заболевания, изоляция больных животных от здорового поголовья.
ВНИМАНИЕ
Приобретая молоко на стихийных рынках или у соседей, не стесняйтесь поинтересоваться, подвергалось ли их животное диагностическим исследованиям за последние полгода и каков был результат. Далеко не все люди кипятят домашнее молоко. Речь идет о вашем здоровье!
Иммунитета у животных на данное заболевание нет, поэтому абсолютно устойчивых пород КРС к лейкозу тоже нет.
Животные не проявляют клинических симптомов заболевания длительное время. Иногда, явные признаки могут вовсе не проявиться за всю жизнь животного на подворье. Корова-вирусоноситель, не проявляя никаких симптомов заболевания, может быть продуктивной и давать большие удои молока. У заражённых лейкозом коров зачастую рождаются здоровые телята, которые впоследствии могут заразиться болезнью от животных-вирусоносителей.Такие явные симптомы лейкоза, как увеличенные лимфоузлы, снижение иммунитета, истощение, увеличенная селезёнка, низкая продуктивность, наступают в терминальной, заключительной стадии заболевания. При вскрытии погибшего, или забитого животного обнаруживается, что весь его организм поражён опухолями. Туша такого животного должна подвергнуться утилизации.
У заражённых лейкозом коров зачастую рождаются здоровые телята, которые впоследствии могут заразиться болезнью от животных-вирусоносителей.Такие явные симптомы лейкоза, как увеличенные лимфоузлы, снижение иммунитета, истощение, увеличенная селезёнка, низкая продуктивность, наступают в терминальной, заключительной стадии заболевания. При вскрытии погибшего, или забитого животного обнаруживается, что весь его организм поражён опухолями. Туша такого животного должна подвергнуться утилизации.
Особенности диагностики
Выявить больное животное можно только путём лабораторного исследования его крови.
Лечение и профилактика лейкоза у коров.
Лейкоз крупного рогатого скота считается неизлечимым.
При выявлении у двух и более животных, с положительным диагнозом на лейкоз – хозяйство считается неблагополучным по этому заболеванию. Ему предписываются ограничения, и разрабатывается план оздоровления стада.
В таком хозяйстве запрещается:
• содержать инфицированных животных совместно со здоровым поголовьем. Они подлежат или забою на мясо, или утилизации;
Они подлежат или забою на мясо, или утилизации;
• использовать в пищу лейкозное молоко;
• перемещать больных животных в пределах населённого пункта без ведома ветеринарной службы;
Требуется полная изоляция животных-вирусоносителей от здорового стада (отдельные помещения, выпас, родильное отделение). При проведении ветеринарно-зоотехнических мероприятий необходимо строго соблюдать правила асептики и антисептики. Также необходимо неукоснительно соблюдать карантинный режим, при увеличении поголовья путем закупки из других фермерских хозяйств.
Оздоровление стада.
Оздоровление хозяйства проводится различными способами, в зависимости от степени зараженности животных лейкозом. Все мероприятия регламентированы «Правилами по профилактике и борьбе с лейкозом КРС».
На данный момент единственный способ оградить здоровое поголовье крупного рогатого скота от заражения лейкозом – выбраковка животных-вирусоносителей из стада.
В конечном итоге все методы сводятся к тому, что хозяйство систематически проверяется на заболевание, проводится убой больных животных и замена стада.
Богдановичский район с января 2002 года официально считается оздоровленным от лейкоза.
Однако сохраняется постоянная угроза заноса заболевания из неблагополучных районов. Периодически нашей ветеринарной лабораторией выявляются животные- вирусоносители, как правило, причиной их появления является несанкционированный завоз крупного скота в личные подсобные хозяйства из соседних областей.
Лейкоз крупного рогатого скота (leukaemia in cattle)
Лейкоз крупного рогатого скота (leukaemia in cattle) – хроническая инфекционная болезнь опухолевой природы, основной причиной которой – злокачественное разрастание клеток кроветворных органов с нарушением их созревания, в результате чего происходи диффузная инфильтрация органов этими клетками или появляются опухоли.
Этиология. Возбудителем инфекции является вирус лейкоза крупного рогатого скота (ВЛ КРС)– Bovine Leukemia virus, относящийся к семейству Retroviridae, подсемейству Oncoviridae типа С.
Эпизоотологические данные. Источник инфекции – больные лейкозом животные. В естественных условиях ВЛ КРС может передаваться пренатально и постнатально. Механизм пренатальной передачи включает передачу вирусного генома через гаметы. Постнатальная передача включает передачу вируса через молоко или при контакте. Контактная передача может быть результатом прямого воздействия контаминированных вирусом секретов или экскретов или переноса вируса насекомыми или контаминированными объектами. Так как частицы ВЛ КРС не продуцируются in vitrо, то в большинстве случаев вирус передается с инфицированными лимфоцитами. Для заражения коровы достаточно ввести внутрикожно 2500 лимфоцитов крови.
Течение и симптомы. Встречается 2 формы лейкоза – энзоотическая и спорадическая. Спорадическая форма лейкоза встречается редко и поражает животных до 3-х летнего возраста.
Встречается 2 формы лейкоза – энзоотическая и спорадическая. Спорадическая форма лейкоза встречается редко и поражает животных до 3-х летнего возраста.
Имеет три формы проявления: ювинальный лейкоз характеризуется увеличением лимфоузлов и часто инфильтрацией костного мозга; тимусная форма отмечается у телят до 2-х летнего возраста и характеризуется увеличением тимуса; кожная форма встречается у телят от 1 до 3-х летнего возраста, характеризуется узелковой лейкемической инфильтрацией кожи. Энзоотический лейкоз – контагиозная болезнь с длительно латентным периодом, во время которого в крови выявляется вирус лейкоза (ВЛ КРС) и антител к нему. В основном встречается у крупных животных в возрасте 5-8 лет.
Болезнь характеризуется пролиферацией неопластических элементов, в результате чего образуются отдельные опухолевые массы или диффузная инфильтрация различных тканей и органов. В опухолевый процесс вовлекаются лимфоузлы, часто поражаются селезенка, сычуг, сердце, почки и другие органы.
Диагноз на лейкоз ставят на основании эпизоотологических данных, клинических признаков, патологоанатомических изменений и результатов лабораторных исследований, которые включают серологические, гематологические, гистологические и вирусологические исследования.
При проведении серологических исследований в сыворотках крови определяют наличие антител в ВЛ КРС в РИД, ИФА. Для проведения вирусологических исследований используют пораженные ткани и лимфоциты, в которых выявляют наличие провирусной ДНК с помощью ПЦР, и элементарных частиц вируса с помощью электронной микроскопии.
Гематологическими исследованиями выявляют повышенное содержание лейкоцитов, в основном лимфоидного ряда, слабодифференцированных лимфоидных клеток.
Дифференциальный диагноз. Лейкоз необходимо отличать от актиномикоза, туберкулеза, паратуберкулеза и бруцеллеза.
Лечение больных лейкозом животных не проводят, их подвергают вынужденному убою.
Профилактика и меры борьбы. Для профилактики лейкоза предложен ряд рекомбинатных вакцин, а также вакцин, основанных на использовании поверхностные антигены вируса gp 51, gp 30, p24. В условиях лабораторий предложенные вакцины оказались эффективными, но пока широкого применения не получили.
Основной метод профилактики и борьбы с лейкозом в различных странах мира – своевременно проведенная диагностика и удаление из стада инфицированных животных. При инфицированности стад вирусом лейкоза свыше 20% инфицированных животных изолируют в отдельное стадо и эксплуатируют с предосторожностью. При инфицированности ВЛ КРС ниже 20% наиболее радикальный способ – выведение их из стада с последующей сдачей на мясокомбинат и замена их здоровыми животными. Но в различных странах имеются свои региональные системы оздоровления от лейкоза, основанные на диагностике и выделении из стада инфицированных животных.
Статьи по теме:
Морфологические и биохимические показатели крови коров, инфицированных влкрс, в условиях биогеохимического субрегиона
Лейкоз КРС
Дата публикации: 20 декабря 2019 г.
Лейкоз крупного рогатого скота – хроническая болезнь опухолевой природы, протекающая бессимптомно или характеризующаяся лимфоцитозом и злокачественным разрастанием кроветворных и лимфоидных клеток в различных органах.
Возбудитель болезни у КРС
Вирус лейкоза крупного рогатого скота – РНК-содержащий вирус подсемейства Oncornavirinae (опухолевые вирусы) семейства Retroviridae. Основным признаком всех представителей семейства Retroviridae является наличие в вирионе обратной транскриптазы. В состав подсемейства Oncornavirinae входят три рода, дифференцируемые на основании морфологии вирионов: вирусы типов С, В и D. Вирусы типа С делят на два подрода: вирусы типа С млекопитающих и вирус лейкоза крупного рогатого скота (ВЛКРС).
ВЛКРС – это экзогенный вирус в отношении крупного рогатого скота и других чувствительных видов животных. Репликация вируса ограничивается клетками лимфоидной популяции и не выявлена в других тканях организма. ВЛКРС репродуцируется в перевиваемых, хронически инфицированных культурах клеток тканей животных разных видов.
Установлено, что ВЛКРС чувствителен к температурным воздействиям, разрушается при повторяющихся замораживаниях и оттаиваниях и при прогревании при 56 «С в течение 15 мин. Пастеризация молока (74 °С в течение 16 с) разрушает ВЛКРС. Полная инактивация вируса в молоке или вируссодержащей жидкости (кровь, молозиво) установлена при 50 «С в течение 70 с, при 70…74 «С – за 17 с. Прямой солнечный свет инактиви-рует ВЛКРС в течение 4 ч, УФ-лучи – в течение 30 мин. Вирус полностью теряет активность в 2%-ных растворах гидроксида натрия и формальдегида и других дезинфектантов в общепринятых концентрациях.
Таким образом, решающее этиологическое значение ВЛКРС при возникновении гемобластозов крупного рогатого скота доказано. Вместе с тем при изучении вирусной этиологии опухолей и лейкозов животных накапливались достоверные данные о том, что канцерогенный эффект вирусов проявляется в зависимости от иммунобиологического состояния организма и воздействия стресс-факторов.
ВЛКРС репродуцируется в перевиваемых, хронически инфицированных культурах клеток тканей животных разных видов.
Установлено, что ВЛКРС чувствителен к температурным воздействиям, разрушается при повторяющихся замораживаниях и оттаиваниях и при прогревании при 56 «С в течение 15 мин. Пастеризация молока (74 °С в течение 16 с) разрушает ВЛКРС. Полная инактивация вируса в молоке или вируссодержащей жидкости (кровь, молозиво) установлена при 50 «С в течение 70 с, при 70…74 «С – за 17 с. Прямой солнечный свет инактиви-рует ВЛКРС в течение 4 ч, УФ-лучи – в течение 30 мин. Вирус полностью теряет активность в 2%-ных растворах гидроксида натрия и формальдегида и других дезинфектантов в общепринятых концентрациях.
Таким образом, решающее этиологическое значение ВЛКРС при возникновении гемобластозов крупного рогатого скота доказано. Вместе с тем при изучении вирусной этиологии опухолей и лейкозов животных накапливались достоверные данные о том, что канцерогенный эффект вирусов проявляется в зависимости от иммунобиологического состояния организма и воздействия стресс-факторов.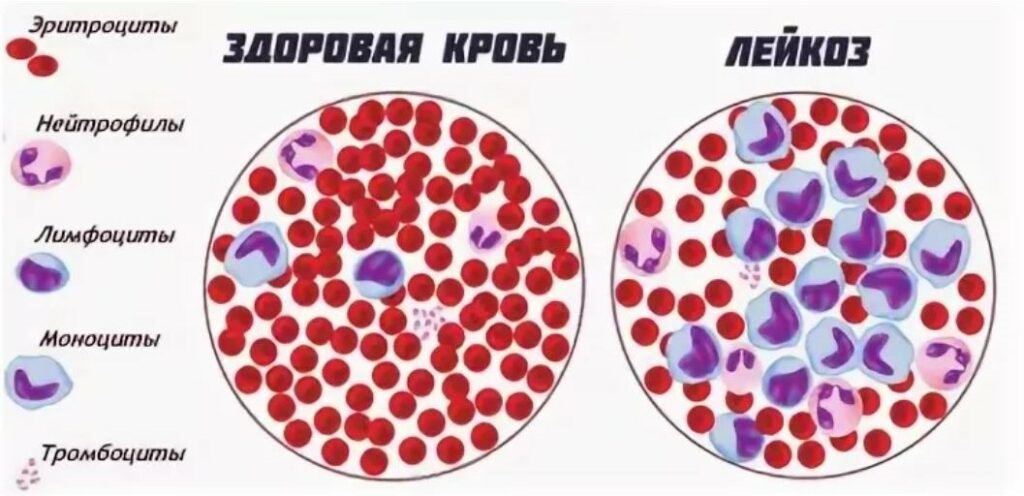 Отмечается также более выраженная генетическая устойчивость отдельных пород и линий животных к ВЛКРС.
В естественных условиях к ВЛКРС восприимчив крупный рогатый скот. Лейкозом болеют молодые и взрослые животные всех разводимых пород и помесей, но чаще эту болезнь отмечают у животных старше 4 лет. Телята до 6-месячного возраста устойчивы к ВЛКРС, что обусловлено, вероятно, колостральным иммунитетом. Уровень инфицированное™ животных в возрасте 6…24 мес также низкий.
Источник возбудителя заболевания у КРС – больные гемобластозами животные. В естественных условиях вирус лейкоза может передаваться пренатально и постнатально. Вирус передается от коровы телятам трансплацентарно во время последних 6 мес внутриутробной жизни. Постнатальная (т.е. горизонтальная) передача включает передачу вируса через молоко или при контакте. В большинстве случаев (более 90%) вирус передается с инфицированными лимфоцитами.
Основным естественным фактором передачи ВЛКРС считается молоко инфицированных и особенно больных на клинико-гематологических и опухолевых стадиях коров.
Отмечается также более выраженная генетическая устойчивость отдельных пород и линий животных к ВЛКРС.
В естественных условиях к ВЛКРС восприимчив крупный рогатый скот. Лейкозом болеют молодые и взрослые животные всех разводимых пород и помесей, но чаще эту болезнь отмечают у животных старше 4 лет. Телята до 6-месячного возраста устойчивы к ВЛКРС, что обусловлено, вероятно, колостральным иммунитетом. Уровень инфицированное™ животных в возрасте 6…24 мес также низкий.
Источник возбудителя заболевания у КРС – больные гемобластозами животные. В естественных условиях вирус лейкоза может передаваться пренатально и постнатально. Вирус передается от коровы телятам трансплацентарно во время последних 6 мес внутриутробной жизни. Постнатальная (т.е. горизонтальная) передача включает передачу вируса через молоко или при контакте. В большинстве случаев (более 90%) вирус передается с инфицированными лимфоцитами.
Основным естественным фактором передачи ВЛКРС считается молоко инфицированных и особенно больных на клинико-гематологических и опухолевых стадиях коров. Учитывая возможность переноса ВЛКРС ничтожным количеством крови, следует иметь в виду, что при несоблюдении правил асептики и антисептики болезнь может распространиться во время проведения ветеринарных и зоотехнических процедур (ятрогенный фактор передачи): ректальные исследования, фиксация за носовые перегородки, бонитировка, массовые вакцинации, взятия крови, проведение хирургических операций и т.п.
По интенсивности развития эпизоотического процесса гемобластозы крупного рогатого скота относятся к медленно развивающимся инфекционным болезням.
Контагиозность лейкоза незначительна. Сезонность не проявляется. Природно-географические и климатические факторы не влияют на распространение болезни у КРС.
У некоторых, чаще всего взрослых животных, несущих наследственную предрасположенность, при дополнительных молекулярно-биологических и иммунобиологических перестройках в клетке болезнь проявляется гематологическими и реже опухолевыми изменениями в органах кроветворения.
Учитывая возможность переноса ВЛКРС ничтожным количеством крови, следует иметь в виду, что при несоблюдении правил асептики и антисептики болезнь может распространиться во время проведения ветеринарных и зоотехнических процедур (ятрогенный фактор передачи): ректальные исследования, фиксация за носовые перегородки, бонитировка, массовые вакцинации, взятия крови, проведение хирургических операций и т.п.
По интенсивности развития эпизоотического процесса гемобластозы крупного рогатого скота относятся к медленно развивающимся инфекционным болезням.
Контагиозность лейкоза незначительна. Сезонность не проявляется. Природно-географические и климатические факторы не влияют на распространение болезни у КРС.
У некоторых, чаще всего взрослых животных, несущих наследственную предрасположенность, при дополнительных молекулярно-биологических и иммунобиологических перестройках в клетке болезнь проявляется гематологическими и реже опухолевыми изменениями в органах кроветворения.
Течение и клиническое проявление болезни у КРС
Лейкозы характеризуются длительным латентным периодом (инкубационный период), во время которого в крови выявляют ВЛКРС и антитела к нему. При спонтанном заражении этот период длится от 2 до 6 лет. Клинические проявления зависят от вовлечения в патологический процесс органов – лимфатических узлов, селезенки, сычуга, сердца, почек, половых и др. Инфекционный процесс при лейкозе развивается медленно и незаметно. В развитии лейкозного процесса у крупного рогатого скота различают четыре стадии: предлейкозную, начальную (доклиническую), развернутую (клинико-гематологическую) и конечную, или терминальную (опухолевую), стадии.
Предлейкозная стадия у зараженных ВЛКРС животных проявляется в виде относительного лимфоцитоза (до 14 тыс/мкл, или 14 – 109/л) за счет лимфоидных клеток, характерных для подозрительных по заболеванию по «лейкозному ключу». Обнаружить другие патологические изменения не удается.
Начальная стадия заболевания КРС характеризуется отсутствием клинических признаков болезни, но более постоянными изменениями количественного и качественного состава крови. Число лейкоцитов колеблется от 15 до 40 тыс/мкл [(15…40) Ю9/л], а среди лимфоцитов преобладают юные и средние клетки.
При спонтанном заражении этот период длится от 2 до 6 лет. Клинические проявления зависят от вовлечения в патологический процесс органов – лимфатических узлов, селезенки, сычуга, сердца, почек, половых и др. Инфекционный процесс при лейкозе развивается медленно и незаметно. В развитии лейкозного процесса у крупного рогатого скота различают четыре стадии: предлейкозную, начальную (доклиническую), развернутую (клинико-гематологическую) и конечную, или терминальную (опухолевую), стадии.
Предлейкозная стадия у зараженных ВЛКРС животных проявляется в виде относительного лимфоцитоза (до 14 тыс/мкл, или 14 – 109/л) за счет лимфоидных клеток, характерных для подозрительных по заболеванию по «лейкозному ключу». Обнаружить другие патологические изменения не удается.
Начальная стадия заболевания КРС характеризуется отсутствием клинических признаков болезни, но более постоянными изменениями количественного и качественного состава крови. Число лейкоцитов колеблется от 15 до 40 тыс/мкл [(15…40) Ю9/л], а среди лимфоцитов преобладают юные и средние клетки. Гематологические изменения могут многие годы оставаться стабильными. При этом общее состояние животного – упитанность, молочная продуктивность и воспроизводительная функция не вызывают подозрений на лейкоз. Лишь при обострении хронического течения болезни могут появляться такие признаки, как снижение удоя, истощение и другие, и лейкозный процесс переходит в развернутую стадию.
Развернутая стадия заболевания КРС характеризуется кроме гематологических сдвигов разнообразием специфических и неспецифических клинических признаков. У животного ухудшается общее состояние, отмечаются быстрая утомляемость, плохой аппетит, снижаются удои, прогрессирует истощение, наблюдается атония преджелудков, сменяющаяся диареей. Перкуссией нередко устанавливают увеличение сердца, число пульсовых ударов достигает ПО… 120 в минуту, пульс слабого наполнения, волна малая. Сердечный толчок ослаблен. При сердечной слабости у животного развиваются отеки подкожной клетчатки в области подгрудка и межчелюстного пространства.
Гематологические изменения могут многие годы оставаться стабильными. При этом общее состояние животного – упитанность, молочная продуктивность и воспроизводительная функция не вызывают подозрений на лейкоз. Лишь при обострении хронического течения болезни могут появляться такие признаки, как снижение удоя, истощение и другие, и лейкозный процесс переходит в развернутую стадию.
Развернутая стадия заболевания КРС характеризуется кроме гематологических сдвигов разнообразием специфических и неспецифических клинических признаков. У животного ухудшается общее состояние, отмечаются быстрая утомляемость, плохой аппетит, снижаются удои, прогрессирует истощение, наблюдается атония преджелудков, сменяющаяся диареей. Перкуссией нередко устанавливают увеличение сердца, число пульсовых ударов достигает ПО… 120 в минуту, пульс слабого наполнения, волна малая. Сердечный толчок ослаблен. При сердечной слабости у животного развиваются отеки подкожной клетчатки в области подгрудка и межчелюстного пространства. Специфическим клиническим признаком лейкозов крупного рогатого скота является прогрессирующее значительное увеличение поверхностных (околоушных, нижнечелюстных, заглоточных, предлопаточных, над-313 выменных и др.) лимфатических узлов. Отмечается симметричное, но неравномерное увеличение их. Лимфатические узлы достигают величины от 5 до 20 см и более, они безболезненные, подвижные, эластичной или плотной консистенции.
У молодняка наряду с увеличением лимфатических узлов в области голодной ямки, поясницы, шеи часто отмечают опухолевидные разрастания зобной железы, обусловливающие затрудненное дыхание. При значительном увеличении селезенки перкуссией устанавливают притуплённый или тупой звук, в некоторых случаях болезненность. Возможен разрыв селезенки, и животное внезапно погибает вследствие внутреннего кровоизлияния. Значительное увеличение некоторых лимфатических узлов, а также опухолевых разрастаний в тазовой и брюшной областях можно установить при ректальном исследовании. У некоторых (3…5% случаев) больных лейкозами животных отмечают экзофтальм (пучеглазие).
Специфическим клиническим признаком лейкозов крупного рогатого скота является прогрессирующее значительное увеличение поверхностных (околоушных, нижнечелюстных, заглоточных, предлопаточных, над-313 выменных и др.) лимфатических узлов. Отмечается симметричное, но неравномерное увеличение их. Лимфатические узлы достигают величины от 5 до 20 см и более, они безболезненные, подвижные, эластичной или плотной консистенции.
У молодняка наряду с увеличением лимфатических узлов в области голодной ямки, поясницы, шеи часто отмечают опухолевидные разрастания зобной железы, обусловливающие затрудненное дыхание. При значительном увеличении селезенки перкуссией устанавливают притуплённый или тупой звук, в некоторых случаях болезненность. Возможен разрыв селезенки, и животное внезапно погибает вследствие внутреннего кровоизлияния. Значительное увеличение некоторых лимфатических узлов, а также опухолевых разрастаний в тазовой и брюшной областях можно установить при ректальном исследовании. У некоторых (3…5% случаев) больных лейкозами животных отмечают экзофтальм (пучеглазие). Терминальная (опухолевая) стадия лейкоза характеризуется дальнейшим развитием патологического процесса и более отчетливым проявлением клинических признаков: резким увеличением лимфатических узлов, которые могут заметно выступать на теле; отеками подкожной клетчатки в области подгрудка, межчелюстного пространства, конечностей. Это приводит к крайнему истощению органов кроветворения, блокаде иммунной системы и заканчивается смертью животного.
У крупного рогатого скота может встречаться кожная форма лейкоза. На теле животного появляются узелковые припухлости диаметром до 2,5 см, хорошо заметные на шее, спине, крестце и бедрах. В течение нескольких недель происходит облысение припухлости, ее поверхность покрывается корочкой, состоящей из эпителия и экссудата. Затем корочки отпадают, а облысевшие участки вновь покрываются шерстью. Однако через несколько месяцев после кажущегося выздоровления наступает рецидив с появлением тех же признаков болезни. Происходит инфильтративное поражение висцеральных органов, и животное погибает.
Терминальная (опухолевая) стадия лейкоза характеризуется дальнейшим развитием патологического процесса и более отчетливым проявлением клинических признаков: резким увеличением лимфатических узлов, которые могут заметно выступать на теле; отеками подкожной клетчатки в области подгрудка, межчелюстного пространства, конечностей. Это приводит к крайнему истощению органов кроветворения, блокаде иммунной системы и заканчивается смертью животного.
У крупного рогатого скота может встречаться кожная форма лейкоза. На теле животного появляются узелковые припухлости диаметром до 2,5 см, хорошо заметные на шее, спине, крестце и бедрах. В течение нескольких недель происходит облысение припухлости, ее поверхность покрывается корочкой, состоящей из эпителия и экссудата. Затем корочки отпадают, а облысевшие участки вновь покрываются шерстью. Однако через несколько месяцев после кажущегося выздоровления наступает рецидив с появлением тех же признаков болезни. Происходит инфильтративное поражение висцеральных органов, и животное погибает.
Диагностика и дифференциальная диагностика болезни у КРС
Первичный диагноз в хозяйстве ставят на основании эпизоотологических, клинико-гематологических, серологических и патологоанатомических данных с обязательным проведением гистологического исследования.
Корову считают больной лейкозом при наличии одного из следующих показателей: 1) клинических признаков болезни; 2) положительных результатов гематологических исследований; 3) обнаружении у павшего (убитого) животного характерных патологоанатомических изменений; 4) установлении положительного результата гистологического исследования патологического материала в случае падежа (убоя) животных.
Клинические признаки проявляются, как правило, к концу болезни, поэтому в диагностике заболевания они имеют лишь вспомогательное значение.
Гематологическое исследование заключается в обнаружении в периферической крови повышенного числа лейкоцитов, в основном лимфоидного ряда, и слабодифференцированных клеток (родоначальных, пролимфоцитов, лимфобластов), а также полиморфных, атипичных клеток
Серологический метод диагностики основан на выявлении в сыворотке крови животных при помощи РИД антител к антигену ВЛКРС. Специфические антитела появляются в крови крупного рогатого скота через 2 мес после заражения, т.е. значительно раньше, чем гематологические изменения, и сохраняются пожизненно. РИД представляет собой основной метод серологического исследования животных в государственных программах по борьбе с лейкозом крупного рогатого скота во многих странах, в том числе в России.
При дифференциальной диагностике следует принимать во внимание ряд хронических инфекционных болезней крупного рогатого скота (актиномикоз, туберкулез, паратуберкулез, бруцеллез), а также некоторые незаразные заболевания (гепатиты, циррозы, амилоидную дистрофию и другие заболевания печени, маститы, пневмонии, ретикулоперикардиты, нефриты), сопровождающиеся лейкозоподобными изменениями гематологических показателей, называемых лейкемоидными реакциями.
Профилактика болезни у КРС
Общие мероприятия по профилактике лейкоза крупного рогатого скота включают в себя соблюдение ветеринарно-санитарных требований при содержании, кормлении и ветеринарном обслуживании животных. Продажу, сдачу на убой, выгон, размещение на пастбищах и все другие перемещения и перегруппировки животных, реализацию животноводческой продукции проводят только с ведома и разрешения ветеринарных специалистов. Осуществляют карантинирование в течение 30 дней вновь поступивших животных для проведения клинического осмотра, серологического и гематологического исследований.
Контроль за благополучием поголовья скота осуществляют на основании показателей послеубойной экспертизы на мясокомбинатах; данных экспертизы при внутрихозяйственном убое животных, вскрытиях трупов животных; результатов плановых серологических и гематологических исследований на лейкоз; результатов контрольного убоя животных с повышенным уровнем лимфоцитов в крови и патоморфологических исследований биоматериалов.
Лечение болезни у КРС
Не разработано.
Лейкоз КРС / Россельхознадзор
Вирусный лейкоз крупного рогатого скота (ВЛКРС) относят к заболеваниям опухолевой природы – гемобластозам, основным признаком которых является злокачественное разрастание клеток кроветворной ткани и нарушение их созревания.
Лейкоз крупного рогатого скота (далее КРС) в настоящее время получил широкое распространение практически во всех регионах нашей страны и за рубежом (Великобритания, Португалия, Филиппины, Италия, Франция, Германия, США, Канада, Турция, Греция, Южная Африка и др.). Так по данным ФГБУ «Федеральный центр охраны здоровья животных», в России треть поголовья КРС заражена лейкозом. Наибольший удельный уровень заболевания приходится на Центральный и Приволжский федеральные округа. В Томской области неблагополучных пунктов по лейкозу КРС 5: в Томском (1), Шегарском (2), Зырянском (1) и Кожевниковском (1) районах.
Мероприятия по профилактике и ликвидации лейкоза КРС проводятся в соответствии с «Правилами по профилактике и борьбе с лейкозом крупного рогатого скота», утвержденными приказом Минсельхозпрода РФ от 11.05.1999 N 359.
При формировании стада необходимо следить за благополучием и эпизоотической ситуацией того хозяйства, из которого производится завоз животных. Не допускать ввоз на территорию животных из неблагополучных пунктов.
Контроль за благополучием поголовья скота осуществляют ветеринарные специалисты хозяйств, государственной ветеринарной службы в результатов плановых серологических и гематологических исследований на лейкоз.
Владельцы, занимающиеся реализацией животных, обязаны обеспечить ежегодное однократное проведение клинических осмотров и серологических исследований всех животных старше 6-месячного возраста, а в остальных хозяйствах контроль за благополучием по лейкозу осуществляют путем ежеквартального клинического осмотра и по результатам ветсанэкспертизы при убое или патолого — анатомическом вскрытии павших животных.
Быки — производители всех категорий хозяйств подлежат исследованию на лейкоз серологическими методами не менее двух раз в год с интервалом 6 месяцев.
Животных, принадлежащих гражданам, проживающим на территории хозяйств или в отдельных населенных пунктах, исследуют на лейкоз одновременно с проведением этой работы на фермах, а также в случаях подозрения на заболевание животных лейкозом.
В благополучных хозяйствах исследования проводят 1 раз в год, быков-производителей — 2 раза в год. Регулярные, серологические исследования позволяют своевременно выявить подозрительных и выделять их из стада.
При выявлении больных животных хозяйство объявляют неблагополучным. Оздоровление проводят путём серологических исследований с последующим удалением больных, либо полной заменой всего стада.
Хочется призвать фермеров не замалчивать проблему лейкоза в своем хозяйстве. Ведь каждый сокрытый случай заболевания коровы лейкозом усугубляет эпизоотическую обстановку района, и в какой-то момент эпидемия может стать неуправляемой. В итоге район теряет стабильность в экономических показателях.
Источник: Управление Федеральной службы по ветеринарному и фитосанитарному надзору по Томской области
Информационная памятка по профилактике лейкоза крупного рогатого скота
Новости Информационная памятка по профилактике лейкоза крупного рогатого скота Лейкоз крупного рогатого скота — это хроническая инфекционная, медленно протекающая болезнь опухолевой природы. Болезнь вызывает опухолевый процесс. О том, что существует такое заболевание, стало известно относительно недавно – в 1969 году. А вообще первое сообщение о лейкозе у КРС было еще в 1871 году. Все это время ветеринары и фермеры практически не имели представления, чем именно болеют животные, потому что внешние признаки заболевания очень размытые, а на начальных стадиях определить ее без специальных анализов часто невозможно. Поэтому много десятилетий заболевание быстро распространялось без каких-либо препятствий.
Почему лейкоз КРС так распространен и как он попал в Россию?
Веками главной целью селекционеров было сделать коров более продуктивными. Остальные характеристики пород отодвигались на второй план. Так получилось, что продуктивность скота росла в ущерб его иммунитета. Достаточно сказать, что у диких животных лейкоз поражает не более 5% особей.
В Россию лейкоз КРС мог попасть вместе завозом высокопродуктивного скота. Точно не известно как заболевание попало в Россию, но некоторые связывают его появление с завозом немецкого племенного скота в 1940 году и затем в 1945-1947 годах. Утверждать что-либо точно невозможно, так как сам вирус был выделен только в 1969 году.
Сама болезнь заключается в появлении злокачественных образований в органах и тканях животного, но прежде всего в кроветворных. Процесс затрагивает лимфоузлы, селезенку, сычуг, сердце, почки и другие органы.
У лейкоза есть стадии заболевания. Первая стадия (также называется инфекционной) проходит без каких-либо внешних проявлений. Более того, сохраняется продуктивность и воспроизводительная функция скота. Симптомы появляются только со второй стадии, которая называется патологической. При ней появляются гематологические изменения в периферической крови (то есть той, которая циркулирует по сосудам вне кроветворных органов). Конечная стадия – самая заметная. Именно на ней начинается снижение продуктивности и воспроизводительной функции, а во внутренних органах заметны нарушения функционального состояния.
Источником инфекции является зараженное животное. Передача вируса происходит через кровь, молоко, биологические жидкости, инфицированные предметы ухода за поголовьем, при воспроизводстве стада от зараженных производителей. Заражение телят происходит в пренатальный (от матери к плоду) период, а также после рождения – через молоко. Заразившись однажды, животное остается инфицированным пожизненно.
Главным фактором распространения является совместное содержание здоровых животных с вирусоносителями лейкоза.
Характерные признаки болезни отсутствуют. Выявить животное вирусоносителя возможно только в ходе лабораторных исследований. У животного в течение длительного периода развивается ухудшение общего состояния, нарастает быстрая утомляемость, ухудшение аппетита, снижение удоя, прогрессирует истощение, наблюдается атония преджелудков, сменяющаяся диареей.
Лечение животных не существует.
С целью ранней диагностики все животные с 6-месячного возраста обязаны подвергаться профилактической диагностике. В случае выявления вирусоносительства животные переводятся в группу откорма. По окончании откорма сдаются на убой. Вирусоносители старше 12-месячного возраста дополнительно подвергаются гематологическим исследованиям. При выявлении гематологических изменений животных без откорма сдаются на убой.
Для предупреждения появления лейкоза в хозяйствах всех форм собственности ОГУ «Балаковская райСББЖ» напоминает о необходимости соблюдения мер профилактики.
Так, владельцам ферм и личных подворий требуется придерживаться следующих правил:
— периодически (раз в 6 месяцев) предоставлять животное ветеринарному специалисту; государственной службы для отбора проб на серологические исследования;
— приобретать животных только при наличии ветеринарных сопроводительных документов в благополучных по инфекционным заболеваниям хозяйствах;
— при покупке КРС извещать государственную ветеринарную службу для всестороннего обследования, включая лейкоз, и постановки животного на карантин;
— не допускать к воспроизводству молодняк, рожденный от инфицированных коров.
Обо всех случаях подозрения на наличие вышеназванного заболевания просим незамедлительно сообщать в ОГУ «Балаковская райСББЖ» по телефонам: 44-13-17; 44-27-29; 44-08-05.
Просмотров: 701
Что-нибудь ближе к настоящему?
Метастаз рака Ред. Авторская рукопись; доступно в PMC 2014 1 июня.
Опубликован в окончательной отредактированной форме как:
PMCID: PMC3568447
NIHMSID: NIHMS416371
Раздел по гематологии и онкологии, Wake Forest Baptist Health, Уинстон-Сейлем, другие статьи в ЧВК, цитирующих опубликованную статью.
Abstract
Модели на животных сыграли неоценимую роль в усилиях по лучшему пониманию и лечению пациентов, страдающих лейкемией.Несмотря на то, что на основе этих моделей были сделаны важные выводы, следует признать ограничения. В этом обзоре мы выделим различные модели лейкемии на животных и опишем их вклад в улучшение понимания и лечения этих видов рака.
Введение
Лейкемии представляют собой разнообразную группу заболеваний, которые приводят к значительной заболеваемости и смертности. В 2011 году у более 44 600 американцев была диагностирована лейкемия, что привело к 21 780 смертельным исходам [1]. Четыре основных типа составляют 85% всех лейкозов: острый миелоидный лейкоз (AML), острый лимфобластный лейкоз (ALL), хронический миелоидный лейкоз (CML) и хронический лимфолейкоз (CLL).Этим и будет посвящен этот обзор. ХЛЛ является наиболее распространенным заболеванием, но на ОМЛ приходится ~ 42% всех смертей от лейкемии [1]. В результате ОМЛ является наиболее интенсивно изучаемым лейкозом и имеет самую разнообразную группу моделей на животных. Трудно переоценить влияние животных моделей на исследования лейкемии. Начиная с новаторской работы доктора Ллойда Лоу о реакции мышиного лимфоидного лейкоза на антиметаболиты в 1940-х и 1950-х годах до открытия лейкемической стволовой клетки доктором Джоном Диком в 1990-х годах, животные модели были неотъемлемой частью нашего понимания биология лейкозов.
Модели ОМЛ на животных
Введение
За последние 40 лет наше понимание молекулярной патофизиологии ОМЛ значительно выросло (обзор в [2–4]). Заболевание проявляется увеличением незрелых, клональных, миелоидных предшественников, что приводит к прогрессирующей недостаточности костного мозга и, в конечном итоге, к смерти. Несмотря на лучшее понимание генетики ОМЛ, терапия для большинства пациентов осталась неизменной, а общая 5-летняя выживаемость составляет 30-40% [5].Этот неутешительный показатель выживаемости вдохновил на большую работу по разработке более подходящих моделей на животных для использования при обнаружении новых целей и тестировании новых методов лечения. В результате несколько исследований выявили новые идеи, которые стали возможными только благодаря использованию моделей на животных. Несколько примечательных примеров включают роль взаимодействия клеток AML / стромы костного мозга в устойчивости к химиотерапии [6], способность иммунной системы взаимодействовать с AML и нацеливаться на них [7-8], манипулирование нишей гемопоэтических стволовых клеток посредством AML [9], а также плодотворное наблюдение, что AML существует как иерархия клеток с редкой популяцией лейкемических стволовых клеток [10].Это наблюдение стало центром теории раковых стволовых клеток, которая теперь распространяется и на многие солидные опухоли (см. Обзор [11]).
Мышиные модели
С 1930-х гг. Мышиные модели наиболее широко использовались для изучения ОМЛ. Первоначальные трансплантируемые модели, индуцированные канцерогенами, уступили место более современным трансгенным, ксенотрансплантатным и мозаичным подходам. В этом разделе будут рассмотрены основные типы мышиных моделей AML.
Канцероген-индуцированные модели
Среди первых описанных моделей мышиного лейкоза были трансплантируемые канцерогенные модели, используемые для тестирования возможных терапевтических агентов (см.).Использование антиметаболитных агентов было впервые протестировано на этих моделях. У них были преимущества, заключающиеся в том, что они могли размножать in vitro и затем вводить их большим когортам реципиентов, у которых впоследствии разовьется лейкемия, что позволило провести испытания многообещающих агентов и графиков. Они были использованы с большим эффектом докторами Скиппером и Шабелем для описания кинетики лейкемогенеза и ответа на терапию, которые остаются основной опорой нашего современного понимания болезни (см. Обзор в [12]).Одной из наиболее широко используемых моделей является линия клеток L1210, выделенная доктором Лоу путем воздействия на мышей DBA / 2 канцерогенного 3-метилхолантрена (3-MC) [13]. Полученный лейкоз был получен от умирающего животного и десятилетиями использовался экспериментально. Дополнительные химически индуцированные линии клеток мышиного лейкоза включают, среди прочего, P388, P1534 и L5178Y (обзор см. В [14]). Однако у этих моделей есть несколько недостатков. Самым сильным из них является тот факт, что патогенез этого лейкоза не имеет отношения к большинству случаев ОМЛ у людей, поскольку у немногих людей лейкоз развивается после воздействия химических агентов.Действительно, по большей части считалось, что некоторые из этих клеточных линий более тесно связаны с лимфоидными злокачественными новообразованиями, чем ОМЛ. Кроме того, генетические основы болезни неизвестны, что ограничивает применимость результатов с использованием этих моделей. Несмотря на эти недостатки, эти модели внесли значительный вклад в лечение ОМЛ, особенно в поддержку использования цитарабина [15], который остается одним из наиболее широко используемых агентов для лечения ОМЛ и ОЛЛ.
Основные методы создания моделей лейкемии на животных.А) Модели, индуцированные канцерогеном. Животные из восприимчивых штаммов подвергаются воздействию канцерогенных химических веществ или ионизирующего излучения. После заражения животные наблюдаются на предмет развития лейкемии. Б) Мозаика, модели транспозонов и вирусов. В мозаичных моделях HSC собирают от мышей-доноров и заражают вирусами, кодирующими интересующий онкоген. Интеграция вируса в геном хозяина приводит к доставке и экспрессии онкогена. В дополнение к доставке онкогенов внедрение вируса может приводить к аберрантной экспрессии генов клеточных протоонкогенов (как показано) или к нарушению клеточного супрессора опухоли, если интеграция происходит внутригенно.Мутагенез транспозонов является результатом аналогичных последствий интеграции транспозонов в геном хозяина. В) Трансгенные модели. Направляющий вектор, который был сконструирован для экспрессии интересующего трансгена, инъецировали или электропорировали в мышиные ES-клетки. ES-клетки, которые интегрировали вектор, затем вводятся в тетраплоидные бластоцисты, которые, в свою очередь, затем вводятся псевдобеременным суррогатным матерям. В качестве альтернативы векторы могут быть непосредственно введены в оплодотворенные зиготы, а затем эти зиготы имплантированы суррогатным матерям.Затем проводят наблюдение за потомством на предмет развития лейкемии. D) Модели ксенотрансплантатов. Первичные образцы пациентов или линии клеток человека можно вводить непосредственно животным-хозяевам с ослабленным иммунитетом. Животные-хозяева часто подвергаются воздействию ионизирующего излучения перед инъекцией образца для подавления остаточной иммунной функции.
Вирусные и транспозонные модели
Одной из самых ранних вирусных моделей ОМЛ является эритробластный ОМЛ, вызываемый вирусами лейкемии Френда, о котором впервые сообщила в 1957 году доктор Шарлотта Френд [16].Она сообщила о присутствии бесклеточного агента, который может серийно передавать эритробластный лейкоз у восприимчивых линий мышей. Позднее было обнаружено, что этот фильтруемый агент представляет собой комбинацию вируса, формирующего очаг селезенки с дефицитом репликации (SFFV), и репликационно-компетентного вируса мышиного лейкоза Friend (MuLV) [17]. SFFV был идентифицирован как причина эритробластного лейкоза [18]; он частично действует путем инсерционного мутагенеза (см.). Интеграция вирусного генома в непосредственной близости от Spi / PU.1 ген приводит к его сверхэкспрессии, управляемой вирусным LTR, что способствует снижению дифференцировки эритробластов, наблюдаемой при заболевании [19]. MuLV был использован для обнаружения новых генов, участвующих в лейкемогенезе AML, в скринингах инсерционного мутагенеза [20]. Дополнительные вирусы лейкемии, индуцирующие AML, были выделены и использованы в скринингах инсерционного мутагенеза с использованием трансгенных моделей, включая вирус MOL4070LTR [21–22]. Вирусно-индуцированный ОМЛ также использовался в доклинической оценке возможных терапевтических средств, в первую очередь на модели спонтанной лейкемии у мышей AKR в результате онкогенного РНК-вируса [23].Помимо вирусно-опосредованного инсерционного мутагенеза, также были разработаны системы на основе транспозонов, в первую очередь система Sleeping Beauty, которая использовалась для идентификации взаимодействующих мутаций [24-25]. Эти модели, индуцированные вирусами и транспозонами, способствовали пониманию лейкемогенеза и идентификации активных терапевтических агентов. Однако их отношение к заболеванию человека сомнительно, поскольку не было получено убедительных доказательств наличия у человека вируса или транспозона, индуцирующего ОМЛ.
Трансгенные модели
ОМЛ характеризуется неслучайными повторяющимися кариотипическими аномалиями, которые, как известно, влияют на прогноз, особенно хромосомные транслокации. Эти транслокации могут быть патогномоничными для определенного подтипа AML, такого как t (15; 17), наблюдаемого при остром промиелоцитарном лейкозе (APL), или могут возникать при различных лейкозах, таких как филадельфийская хромосома, наблюдаемая при CML, ALL и редко AML.
Самые ранние трансгенные модели мышей, созданных с помощью AML, для экспрессии слитых белков, генерируемых этими транслокациями.В трансгенных моделях мышей создаются путем манипуляции с эмбриональными стволовыми (ES) клетками. В классическом подходе ДНК вводится непосредственно в про-ядро оплодотворенных зигот, которые затем вводятся псевдобеременным самкам (см.). Это приводит к нецелевой интеграции трансгена и зависит от экспрессии трансгена для генерации фенотипа. В более современном подходе ДНК электропорируется в ES-клетки, а интегранты отбираются путем экспрессии генов устойчивости к антибиотикам.Выбранные клетки затем вводятся в тетраплоидные бластоцисты, которые, в свою очередь, имплантируются псевдобеременным самкам. Затем потомство подвергают обратному скрещиванию с мышами дикого типа для получения гомозиготно трансгенных мышей. Теперь можно создать векторы, которые нацелены на определенные сайты в геноме посредством гомологичной рекомбинации. Дополнительный уровень сложности может быть добавлен с помощью более новых векторов, которые условно экспрессируют гены в ответ на доксициклин (системы включения / выключения Tet) или рекомбиназу Cre (с использованием систем Lox / Cre).Как только мыши с этими условными аллелями созданы, их можно скрещивать с другими трансгенными животными, которые экспрессируют трансактиватор Tet или рекомбиназу Cre тканеспецифическим образом, чтобы генерировать тканеспецифическую экспрессию трансгена (обзор в [26]). В дополнение к экспрессии онкогенных слитых белков, онкогенные аллели с усилением функции могут быть «сбиты» с их соответствующих нормальных локусов, а опухолевые супрессоры могут быть «выбиты» с использованием тех же подходов.
APL
Ранние модели APL были созданы путем экспрессии гибридного белка, генерируемого t (15; 17), PML-RAR, под контролем различных миелоид-специфичных промоторов [27–29].Эти модели не только воспроизводили гистологический фенотип APL человека, но также ремиссии, наблюдаемые после лечения всей транс-ретиноевой кислотой (ATRA). В трансгенной модели, экспрессирующей слитый белок PLZF-RAR, обнаруженной у пациентов с редким вариантом APL, содержащим t (11; 17), лечение ATRA не вызывало ремиссии у мышей, имитируя клинический сценарий [30]. Несмотря на эти успехи, у модели есть несколько недостатков. Результирующий фенотип зависит от экспрессии встроенного трансгена.При использовании различных миелоид-специфичных промоторов некоторые промоторы давали модели с высокой пенетрантностью и более короткими латентными периодами, в то время как другие выявляли заболевание, более напоминающее миелопролиферативные новообразования, чем острый лейкоз. Действительно, модель, экспрессирующая PML-RARα под контролем промотора CD11b, вообще не развивала лейкоз [27]. Эти модели все еще оставляют вопрос о том, необходимы ли генетические события в дополнение к t (15; 17), чтобы вызывать APL у людей.
AML1-ETO Leukemias
Слитый белок AML1-ETO генерируется t (8; 21), обнаруживаемым преимущественно в AML FAB класса M2.Пациенты с ОМЛ, положительные по транслокации t (8; 21), имеют лучший прогноз и, как правило, поддаются лечению химиотерапевтическими препаратами [31–32]. Слитые гены AML-ETO приводят к гибели эмбрионов при вставке в эндогенный промотор AML1 из-за плохого гематопоэза, аналогично AML1 — / — животных с нокаутом [33]. Это делает стандартные модели AML1-ETO неинформативными. Чтобы обойти это ограничение, доктор Чжан и его коллеги создали индуцибельную трансгенную модель, которая экспрессирует AML1-ETO под контролем репрессора Tet.Несмотря на устойчивую экспрессию трансгена в костном мозге мышей, лейкоз не развился [34]. Впоследствии они обнаружили, что когда мышей, экспрессирующих AML1-ETO (под контролем миелоид-специфического промотора MRP8 ), лечили канцерогеном, N-этил-N-нитрозомочевиной, у 55% животных развился AML [35], в то время как ни один из них не развивался. контрольных мышей. Это открытие согласуется с мнением, что экспрессия AML1-ETO необходима, но недостаточна для лейкемогенеза. В соответствии с этой идеей другие группы обнаружили сотрудничество AML1-ETO с мутациями в тирозинкиназах, таких как TEL-PDGFR, используя мозаичные модели, которые будут обсуждаться ниже [36–37].
MLL Leukemias
Ген смешанной лейкемии (MLL) расположен на хромосоме 11 и часто участвует в сбалансированных транслокациях при остром лейкозе. У него более 40 известных партнеров слияния при остром лейкозе, и он часто ассоциируется с химиорезистентностью и плохим прогнозом [31–32, 38]. Примерно 5% пациентов с острым лейкозом экспрессируют слияния MLL [39–40], хотя это число увеличилось до ~ 70% пациентов, ранее подвергавшихся воздействию антрациклинов и ядов топоизомеразы II.Модели слияния MLL-белков, таких как Mll-lacZ, приводят к AML у одних животных, но не у других. LacZ не имеет известной онкогенной роли, поэтому это предполагает, что одного MLL достаточно, чтобы вызвать онкогенез. В сценарии t (9; 11) AF9 сотрудничает с MLL. Белок AF9 гомологичен продукту ENL из 19p13.3, который является другим геном-партнером для MLL, участвующего в острых лейкозах. Corral et al. использовали гомологичную рекомбинацию в ES-клетках для получения слияния Mll-AF9. Слитый белок, который они создали, зависит от эндогенных контрольных элементов MLL для транскрипции слитого белка.У мышей развился ОМЛ, несмотря на широко распространенную активность промотора MLL [41]. У небольшой группы животных развился ОЛЛ, что согласуется с тем, что наблюдается у людей [42]. Инженерия реципрокной транслокации между хромосомами 9 и 11 для генерации слитого гена MLL-AF9 была создана в одной модели с использованием сайтов LoxP как в локусах MLL, так и в AF9, но у этих мышей не развился лейкоз [43]. Модель продемонстрировала возможность использования систем Lox-Cre для создания транслокаций у мышей.Другая рекуррентная транслокация с участием гена MLL происходит с t (11; 19) и приводит к генерации слитого белка MLL-ENL. Это событие слияния было смоделировано на трансгенных животных с использованием подхода инженерной транслокации [44]. В этом исследовании мышей получали с сайтами LoxP как в генах MLL, так и в генах ENL и скрещивали с мышами, экспрессирующими Cre с гена Lmo2 , специфичного для гематопоэтических клеток. Полученные в результате мыши развили агрессивный, полностью пенетрантный AML, демонстрируя, что подход инженерной транслокации может успешно моделировать AML.
Модели усиления функции, «сбитые с толку»
Несколько онкогенов вовлечены в развитие ОМЛ у людей, в первую очередь тирозинкиназа рецептора Flt3. Мутации Flt3 встречаются у 25% пациентов с ОМЛ и имеют худший прогноз [45]. Наиболее распространенная мутация Flt3 включает в себя создание тандемной дупликации в рамке считывания, так называемая мутация «Flt3 ITD». Flt3 ITD является конститутивно активной формой Flt3, которая вызывает лиганд-независимую передачу сигналов. Была сгенерирована встроенная модель ITD Flt3; у полученных мышей развилось миелопролиферативное новообразование, но не AML; согласуется с другими моделями, которые предполагают, что ITD Flt3 может приводить к усилению пролиферации, но одного недостаточно для развития AML [46].Также согласуется с этим открытием, когда трансгенных мышей, экспрессирующих ITD Flt3, скрещивали с трансгенными животными, экспрессирующими слитый белок NUP98-HOXD13, обнаруженный у пациентов с миелодиспластическим синдромом (МДС) и AML, в результате у полученного потомства развился высокопенетрантный и летальный AML [47].
G-белок Ras является мишенью для активации мутаций при AML. K-ras мутирует примерно в 10–15% AML и в 20–25% N-ras. Была разработана индуцибельная модель K-ras, управляемая его эндогенным промотором [48].В этой модели исследователи создали мутированный аллель K-ras ниже транскрипционной стоп-последовательности, фланкированной сайтами LoxP. Затем эту мышь скрестили с мышью, которая экспрессирует рекомбиназу Cre под контролем интерферон-индуцируемого промотора MX-1 (MX1-Cre). Условная экспрессия K-ras G12D приводит к фатальному миелопролиферативному нарушению, аналогичному таковому в моделях ITD Flt3. Этот вывод согласуется с гипотезой «двух ударов», согласно которой необходимы два изменения, чтобы вызвать ОМЛ [2].Интересно, что когда аналогичная модель была построена с использованием N-ras G12D , был индуцирован гораздо более вялый миелопролиферативный ответ, и мыши умерли от ряда гематологических злокачественных новообразований, демонстрируя, что разные аллели Ras не являются функционально эквивалентными [49].
Наиболее частая мутация при ОМЛ находится в гене неуклофосмина 1 или NPM1 . Он выполняет множество клеточных функций и перемещается между ядерными, ядрышковыми и цитоплазматическими локациями. При AML ~ 35% пациентов имеют мутацию NPM1, которая изменяет клеточную локализацию, что приводит к цитоплазматическому распределению только мутантного белка.Первая модель, которая сбивала мутированную форму NPM1 в эндогенный локус мыши, привела к легкому миелопролиферативному нарушению, но не к AML. Во второй модели наиболее распространенная форма мутации NPM1 была вставлена в локусы мыши, фланкированные сайтами LoxP; Полученные мыши были скрещены с мышами MX1-Cre. После индукции Cre примерно у одной трети двойных трансгенных мышей развился ОМЛ с отсроченным началом. Пенетрантность увеличивалась, а латентность сокращалась с использованием подхода инсерционного мутагенеза на основе транспозонов.Эти модели прояснили роль мутаций, которые приводят к онкогенному усилению функции в развитии AML, и, вероятно, будут продолжать вносить вклад в более глубокое понимание молекулярного патогенеза заболевания.
«Нокаутированные» модели с потерей функции
Некоторые пути роста и выживания могут быть гиперактивированы из-за потери отрицательных регуляторов. Одним из таких примеров при AML является путь киназы PI3, который гиперактивируется у 50–70% пациентов с AML [50].Супрессор опухолей PTEN является негативным регулятором этого пути. Когда были получены трансгенные мыши, которые содержали сайты LoxP, фланкирующие PTEN, и этих животных скрестили с штаммом, специфичным для миелоида Cre, у 11 из 18 потомков развился AML. Полученный AML был моноцитарным по морфологии, а путь киназы PI3 был активирован во многих случаях экстрамедуллярного моноцитарного AML [51]. Негативным регулятором пути Ras является ген NF1 , который стимулирует ГТФазную активность белков Ras, приводя к их инактивации.Потеря NF1 была спроектирована путем конструирования локусов NF1, фланкированных сайтами LoxP, когда этих животных скрещивали с индуцибельной моделью K-Ras G12D , описанной выше. При индуцировании Cre образовывался агрессивный AML с высокой пенетрантностью [52]. Эти модели демонстрируют, что трансгенный подход может применяться в сочетании с системами LoxP-Cre для моделирования потери опухолевых супрессоров при генерации AML.
Мозаичные модели
Как обсуждалось выше, хотя мышиные трансгенные модели внесли большой вклад в наше понимание AML, эти модели имеют существенные недостатки.Количество времени и усилий, необходимых для перехода от конструирования переносчиков к скринингу мышей-основателей и поддержанию размножающейся колонии, может быть непомерно высоким. Альтернативный подход к трансгенной модели состоит в том, чтобы взять гемопоэтические стволовые клетки (HSC) от мышей, манипулировать ими ex vivo и трансплантировать их аутологичным или сингенным реципиентам (см.). Эта модель позволяет быстро генерировать генетически определенные лейкозы, чаще всего за счет ретро- или лентивирусной трансдукции клеток (см. Обзор [26]).
Векторы на основе вируса стволовых клеток мыши (MSCV) являются наиболее часто используемыми ретровирусами с дефицитом репликации при создании моделей мозаичного лейкоза. Эта быстрая и экономичная система широко использовалась при ОМЛ в дополнение к исследованиям, проведенным на трансгенных животных. Например, исследователи ретровирусно трансфицировали и трансдуцировали гибридный белок AML1-ETO в HSC для получения химерных мышей с молекулярными аномалиями, схожими с таковыми у пациентов с этой транслокацией [53].Модели трансплантации с использованием костного мозга, трансдуцированного с помощью AML1-ETO и N-ras G12D , в сингенных реципиентов, показали сотрудничество, ведущее к лейкемии, которая напоминала подтип M2 AML, наиболее часто связанный с AML1-ETO [31]. Мозаичная модель AML, созданная слиянием MLL-ENL и N-ras G12D , напоминала подтип M4-M5, связанный со слияниями MLL. Когда лейкозных мышей лечили цитарабином и антрациклиновой химиотерапией, ответы на две модели значительно различались: мыши MLL-ENL были плохо восприимчивы, в то время как мыши AML-ETO показали высокие показатели ремиссии и даже несколько излечений.Эти ответы отражают клиническую ситуацию и демонстрируют, что эти модели могут резюмировать различные прогностические эффекты этих транслокаций. Кроме того, мозаичная модель использовалась для моделирования прогностических эффектов мутировавших тирозинкиназ. При использовании для изучения ответа на стандартные химиотерапевтические препараты AML, Flt3 ITD ускорял приживление. Эта модель была чувствительна к цитарабину, обеспечивая снижение опухолевой нагрузки и улучшение выживаемости. Доксорубицин не показал такой же эффективности, и было высказано предположение о чистой резистентности при сочетании двух препаратов, как при стандартной индукционной терапии ОМЛ [54].
Модели ксенотрансплантатов
Даже самый клинически агрессивный образец лейкозных клеток от пациента вряд ли вырастет при помещении в культуру, а характеристики клеток могут измениться в зависимости от используемой среды [55–56]. Кроме того, системы культивирования не могут воспроизводить взаимодействия между лейкозными клетками и их стромой и иммунными клетками. Чтобы обойти эти ограничения, были разработаны мышиные модели ксенотрансплантатов, позволяющие приживить первичные образцы пациентов и клеточные линии.
В первых попытках ксенотрансплантации ОМЛ мышам с ослабленным иммунитетом использовались бестимусные голые мыши, несущие мутацию в гене Foxn1 , что привело к почти полному отсутствию функциональных Т-клеток. Ранние эксперименты с использованием первичных образцов пациентов действительно привели к образованию гранулоцитарных сарком, но без вовлечения костного мозга и других органов [57]. В попытке улучшить приживление костного мозга мышей с тяжелым комбинированным иммунодефицитом (SCID) тестировали на их способность приживать образцы пациентов с AML.У этих мышей есть мутации в гене Prkdc и, следовательно, отсутствуют функциональные Т- или В-клетки. Хотя образцы, введенные внутрибрюшинно или имплантированные в капсулы почек, имели значительную скорость отбора, внутривенная инъекция привела к приживлению очень небольшого количества образцов у животных-реципиентов [58]. В важной статье доктор Джон Дик и его коллеги вводили первичные образцы пациентов мышам SCID, которых затем лечили фактором стволовых клеток человека (SCF) и фактором, стимулирующим колонии гранулоцитов-макрофагов (GM-CSF).Это привело к высокому количеству заборов периферической крови или костного мозга пациента, введенных внутривенно; что еще более важно, только часть лейкозных клеток могла успешно прижиться [10]. Это было первое свидетельство иерархии лейкозных клеток, при которой меньшинство населения отвечает за поддержание болезни. С тех пор были выведены мыши с более полным иммунодефицитом. У мышей с диабетом без ожирения (NOD) / SCID нет функциональных B- или T-клеток и снижена активность NK-клеток и моноцитов.У них более высокая скорость приживления первичных образцов пациентов, чем у мышей SCID [59]. Дальнейшее ослабление иммунитета было установлено у мышей NOD / SCID, у которых есть делеции в гене, кодирующем гамма-цепь рецептора интерлейкина 2 (IL2Rγ) (так называемые мыши NSG). Наконец, на этой платформе была создана трансгенная мышь, которая экспрессирует человеческий iL3, GM-CSF и SCF (NSG-tg). Эта мышь значительно улучшила скорость приживления первичных образцов пациентов по сравнению со стандартной мышью NSG [60]. Хотя возможности этих моделей ограничены в отношении взаимодействий лейкозных иммунных клеток, они являются ценными инструментами для оценки доклинической активности многообещающих агентов и взаимодействий лейкозных клеток со стромой.
Немышиные модели
Для изучения ОМЛ использовались другие животные модели, включая крыс. В то время как спонтанные лейкемии у крыс редки, сообщалось о многих моделях, вызванных канцерогенами и радиацией [61–64]. Миеломоноцитарный лейкоз, L5222, был индуцирован в 1967 г., через 326 дней после однократной внутривенной инъекции этилнитрозомочевины (200 мг / кг массы тела) трехмесячной самке крысы BD IX. Этот лейкоз был трансплантирован крысам BD IX и использовался в доклинических исследованиях эффективности [65].Модель APL, созданная на крысах Brown Norway (BNML), использовалась в доклинических исследованиях химиотерапии и трансплантации [66]. Лейкоз был индуцирован у самок крыс линии BN 9,10-диметил-1,2-бензантраценом [67] и имеет характеристики образования колоний, аналогичные характеристикам образования человеческих образцов AML в анализах колоний [68]. Кроме того, эта модель внесла важный вклад в понимание минимальной остаточной болезни при ОМЛ (см. Обзор [69]). Совсем недавно появились сообщения о системах AML, не относящихся к млекопитающим.Одним из примеров является недавний отчет о трансплантации образцов AML человека эмбрионам рыбок данио. В этом исследовании исследователи вводили человеческие клеточные линии AML и образцы пациентов в эмбрионы рыбок данио через 48 часов после оплодотворения. Они использовали от 50 до 200 клеток и увидели снижение лейкемической нагрузки, когда эмбрионы, которым вводили чувствительные клетки, обрабатывали иматинибом [70]. В другом исследовании человеческие APL и CML в клеточных линиях миелоидного бластного кризиса вводили эмбрионам рыбок данио; Затем эмбрионы обрабатывали иматинибом или ATRA, и у них отмечалось снижение лейкемической нагрузки [71].
Другой модельной системой, используемой для изучения AML, является плодовая муха, Drosophila melanogaster . Экспрессия слитого белка AML1-ETO в клетках крови Drosophila нарушала дифференцировку клеток крови, которые полагаются на RUNX1, подобно тому, что наблюдается у пациентов [72]. Эти системы, не относящиеся к млекопитающим, обладают преимуществами более низкой стоимости и повышенной репродуктивной способности в системах с хорошо изученной генетикой. Они могут внести значительный вклад в понимание патогенеза ОМЛ.
Модели ВСЕ на животных
ВСЕ — наиболее распространенная лейкемия у детей, а также встречается у значительного числа взрослых. Для него характерно агрессивное разрастание клональных лимфобластов. ОЛЛ имеет несколько подтипов, включая пре-В-клетки, Т-клетки и зрелые В-клетки или клетки Беркитта. Он может проявляться в основном в виде твердой массы (лимфобластные лимфомы) или с первичным поражением костного мозга (лимфобластные лейкемии). В отличие от AML, сейчас считается, что более 90% детей с диагнозом ALL могут быть излечены [73].Хотя это замечательное достижение является результатом многих направлений исследований, несколько важных открытий в отношении ALL было обнаружено с использованием моделей на животных. Эти выводы включают демонстрацию того, что активация Notch-1 приводит к Т-клеточному ОЛЛ на мышиной модели трансплантата [74], открытие, что у трансгенных мышей, экспрессирующих версию слияния BCR-ABL p190, развился ОЛЛ [75], и доклиническую оценку активность антиметаболитов [76].
Мышиные модели
Как и в случае AML, мышиные модели ALL используют канцероген-индуцированный, вирусно-индуцированный, трансгенный, мозаичный и ксенотрансплантный подходы.Эти модели внесли свой вклад в нынешнее понимание ВСЕГО.
Модели, индуцированные канцерогенами
Как и в случае AML, ранние исследования на мышах основывались на модели, индуцированной канцерогеном. В дополнение к важной клеточной линии L1210, обсужденной выше, существует несколько других канцероген-индуцированных линий ALL, включая клеточную линию L5178Y. Эта клетка является производной DBA / 2-производной метилхолантрен-индуцированной Т-клеточной линии, которая широко использовалась в доклинических исследованиях эффективности и механизмов устойчивости [77–79].
Вирусные модели
Штамм мышей AKR, склонный к лейкемии, был использован для характеристики онкогенных свойств различных вирусов. В результате была получена модель Т-клеточного ОЛЛ у мышей AKR, инфицированных рекомбинантным ретровирусом, нацеленным на тимоциты [80]. Эта модель использовалась для оценки влияния ожирения на прогрессирование ОЛЛ [81]. Кроме того, лимфобластные лейкозы и лимфомы, возникшие в результате мутагенеза вставки ретровируса, позволили охарактеризовать новые гены, участвующие в лимфоидном лейкемогенезе, путем характеристики общих последовательностей вставки вируса [82].Надежность методов была подтверждена, когда было обнаружено, что идентифицированный с помощью скрининга ген Prdm14 сверхэкспрессируется в человеческом B- и T-клеточном ОЛЛ и может инициировать ОЛЛ при сверхэкспрессии вирусной трансдукцией клеток костного мозга мыши [83].
Трансгенные модели
Было опубликовано несколько ВСЕ трансгенных моделей. Эти модели воспроизводят В-клетки, Т-клетки и лейкоз Беркитта / лимфобластные лимфомы.
BCR-ABL
Слитый белок BCR-ABL является результатом транслокации t (9; 22) (q34; q11), также известной как филадельфийская хромосома.Эта транслокация характерна для ХМЛ, но также обнаруживается при В-клеточном ОЛЛ. Существует несколько изоформ этого слитого белка, форма p210 (обнаруживается в основном при CML) и форма p190 (обнаруживается в основном при ALL). Были созданы трансгенные мыши, экспрессирующие изоформу p190 с промотора BCR, но у них была эмбриональная летальность [75]. Напротив, трансгенные мыши, экспрессирующие слияние p190 BCR-ABL с промотора металлотионеина, рождаются живыми и 95% умирают от пре-B-клеточного лейкоза / лимфомы через 35–200 дней [84].Эта модель была использована для тестирования активности новых соединений [85], ингибиторов тирозинкиназы [86] и влияния ожирения на прогрессирование ОЛЛ [81].
MLL Leukemias
Как следует из названия, лейкемия смешанного происхождения, транслокации MLL наблюдаются как при ОМЛ, так и при ОЛЛ. Слитый белок MLL-AF4 наблюдается у пациентов с t (4; 11), и он сильно связан с В-клеточным ОЛЛ. Была создана модель трансгенной мыши, которая экспрессировала MLL-AF4 с эндогенного промотора MLL. Полученные мыши продемонстрировали нерегулируемый лимфоидный и миелоидный рост, а после длительного латентного периода — В-клеточные лимфомы [87].Другая модель была создана с использованием инвертированного аллеля AF4, нацеленного на интрон в гене MLL; при воздействии рекомбиназы Cre происходила инверсия и образование MLL-AF4. Затем мышей скрещивали с различными трансгенными мышами с тканеспецифической экспрессией Cre. Когда присутствовал Cre, у мышей развивались злокачественные новообразования В-клеток с различной латентностью в зависимости от промотора Cre [88]. Трансгенная модель с использованием слитого белка MLL-AF4 человека, экспрессированного из вирусного LTR MSCV, привела к В-клеточным новообразованиям, и латентность могла быть сокращена путем скрещивания этих животных с трансгенными мышами, экспрессирующими K-ras G12D [89].Этот результат подтверждает гипотезу множественного попадания в лейкемогенез.
Eµ-myc
Лимфома / лейкемия Беркитта считается подтипом ОЛЛ и характеризуется транслокациями, в результате которых ген MYC экспрессируется под контролем промоторов тяжелой или легкой цепи иммуноглобулина. В результате болезнь характеризуется высокой скоростью распространения и очень агрессивным поведением. Были получены трансгенные мыши с мышиным геном Myc под контролем промотора тяжелой цепи IgG, имитирующего t (8; 14), наблюдаемый в клинических условиях.В результате у мышей развились агрессивные В-клеточные лимфомы и лейкозы, при этом 90% животных умерли в течение первых 5 месяцев жизни [90]. Эта модель была использована для изучения роли взаимодействующих мутаций в лимфоме / лейкемогенезе [91], а также в основополагающей статье о том, как потеря p53 или избыточная экспрессия BCL2 влияет на химиотерапевтический ответ in vivo [92].
NOTCh2
Рецепторы Notch — это трансмембранные рецепторы, которые при активации лигандом расщепляются на внеклеточные и внутриклеточные части.Внутриклеточная часть (ICN) перемещается в ядро, где она взаимодействует с дополнительными белками, что приводит к транскрипционной активации генов-мишеней (обзор в [93]). NOTCh2 был впервые идентифицирован при транслокации, несущей ALL, Т-лимфоциты человека, t (7; 9) (q34; q34.3). Эта транслокация приводит к образованию гена слияния, в котором промотор TCR управляет экспрессией 3 ’конца локуса NOTCh2 , кодируя только ICN и приводя к конститутивной активации NOTCh2 [94].Эта транслокация обнаруживается только примерно в 1% Т-клеточного ОЛЛ. Однако в последующем сообщении от 50 до 60% образцов Т-клеточного ОЛЛ содержали точечные мутации в NOTCh2, , причастные к патогенезу большинства типов Т-клеточного ОЛЛ [95]. Было создано несколько моделей трансгенных мышей для экспрессии активированных аллелей NOTCh2 из Т-клеточных промоторов [96–98]. У этих мышей изменено развитие тимоцитов CD4, CD8 и развиваются злокачественные опухоли Т-клеток. В более поздней модели используется трансгенная мышь, которая условно экспрессирует две общие точечные мутации в NOTCh2 из своего эндогенного промотора с использованием кассеты Lox-Stop-Lox.Эти мыши продемонстрировали ускоренное развитие злокачественных опухолей Т-клеток в системе мутагенеза транспозонов Спящей красавицы [99].
Mosaic Models
Как и в случае AML, ex-vivo манипуляции с HSC были использованы для создания мышиных моделей ALL. MSCV-векторы, сконструированные для экспрессии слитого белка p190 BCR-ABL, широко использовались для создания моделей ALL B-клеток, положительных по филадельфийской хромосоме. Несколько примеров включают исследование эффектов химиотерапии на развитие мутантов BCR-ABL, устойчивых к ингибиторам тирозинкиназы (TKI) [100], роль BCL6 в устойчивости к TKI [101] и роль супрессора опухолей Arf. в развитии В-клеточного ОЛЛ [102].Онкогены слияния MLL также широко использовались для создания мозаичных ВСЕХ моделей. Исследователи объединили мозаичную модель с моделью ксенотрансплантата и, используя человеческие HSC, полученные из пуповинной крови, инфицировали их вектором, экспрессирующим MLL-ENL на основе MSCV, и вводили их сублетально облученным мышам NOD / SCID. У 26 из 29 мышей, которым вводили инъекцию, развился агрессивный В-клеточный ОЛЛ, демонстрирующий, что MLL-ЭНЛ может индуцировать ОЛЛ в клетках человека [103]. В дополнительном исследовании слияние MLL-AF9 могло продуцировать AML или ALL в CD34 + клетках пуповинной крови человека в зависимости от мыши-реципиента (NSG против NSG-tg) [104].Эти исследования установили возможность создания моделей ОЛЛ из человеческих клеток, размножающихся у мышей с ослабленным иммунитетом, и начали разгадывать, каким образом лейкозы с перегруппировкой MLL могут быть миелоидными или лимфоидными.
Модели ксенотрансплантатов
Те же линии мышей с ослабленным иммунитетом, описанные в разделе ксенотрансплантатов в моделях AML, были использованы для проведения доклинических исследований эффективности во ВСЕХ образцах. Кроме того, они распространили модель лейкозных стволовых клеток на ОЛЛ, наблюдая, что не все В- или Т-клеточные ОЛЛ-клетки могут инициировать лейкемию у мышей NOD / SCID или NSG [105–106].
Немышиные модели
Подразделение инбредных крыс Sprague-Dawley имеет высокую частоту Т-клеточных злокачественных новообразований и использовалось в доклинической оценке терапевтических средств [107–108]. В-клеточный ОЛЛ был получен от инбредных морских свинок и был серийно трансплантирован инбредным реципиентам, но не беспородным животным [109]. Существуют также модели ОЛЛ, не относящиеся к млекопитающим, включая трансгенную модель рыбок данио, основанную на экспрессии слитого белка TELAML1, обнаруженного у пациентов с пре-В-клеточным ОЛЛ с t (12; 21) (p13; q22).Исследователи экспрессировали трансген либо повсеместно (с использованием модифицированного промотора актина), либо ограниченным лимфоидом (с использованием промотора Rag2). Только у рыбок данио, которые экспрессировали трансген повсеместно, развивался ОЛЛ после длительного латентного периода [110]. Скрининг химического мутагенеза с использованием трансгенных рыбок данио, которые экспрессируют усиленный зеленый флуоресцентный белок Т-клеточно-специфическим образом, дал несколько линий, которые имеют наследственный Т-клеточный ОЛЛ [111]. Другая модель ОЛЛ Т-клеток была создана у рыбок данио с использованием мышиного гена c-myc , экспрессируемого под контролем промотора Rag2 рыбок данио.У трансгенных рыб развился Т-клеточный ОЛЛ с короткой латентностью [112]. В элегантной модели доктор Лук и его коллеги использовали трансгенных рыбок данио, которые содержали условный аллель Cre, управляемый промотором теплового шока 70, и скрестили его с трансгенной моделью, содержащей мышиный c-myc , управляемый промотором Rag2 , содержащим промежуточная кассета Lox-stop-DS Red-Lox. Полученное потомство можно было поместить в воду при 37 ° C, чтобы вызвать экспрессию Cre. Иссечение стоп-кассеты может сопровождаться флуоресцентной визуализацией для обнаружения потери DS Red и индукции экспрессии EGFP.У этих животных развилась Т-клеточная лимфобластная лимфома (LBL), экспрессирующая EGFP, которая после лечения тепловым шоком прогрессировала до лейкемии [113]. Эта модель позже была использована для исследования генетических событий, которые приводят к превращению LBL в ALL [114]. У рыбок данио хорошо зарекомендовавшая себя генетика, низкая стоимость и высокие показатели воспроизводства, что делает их идеальной системой для проведения генетических скринингов. Вклад этих моделей только начинается.
Животные модели CML
ХМЛ уникален среди лейкозов тем, что существует единственное онкогенное событие, которое вызывает заболевание — слитый белок BCR-ABL, генерируемый филадельфийской хромосомой.Это была первая известная соматическая хромосомная аномалия, напрямую связанная со злокачественным новообразованием, и стало первым доказательством того, что рак является генетическим заболеванием [115]. Заболевание характеризуется аномальной пролиферацией и накоплением зрелых и созревающих миелоидных клеток. Он имеет три различных клинических фазы: хроническую фазу, ускоренную фазу и фазу взрыва. В прошлом ХМЛ можно было вылечить только трансплантацией аллогенных стволовых клеток, но в последние годы лечение произвело революцию с появлением ингибиторов киназы BCR-ABL, таких как иматиниб, нилотиниб и дазатиниб (обзор в [116]).
Животные модели сыграли уникальную роль в исследованиях ХМЛ. Доктор Ван Эттен показал, что изоформа p210 белка BCR-ABL непосредственно вызывает ХМЛ, используя мозаичную модель мыши. Он использовал ретровирусный вектор, который экспрессировал белок p210 и инфицированные мышиные HSC. Когда инфицированные клетки были трансплантированы сингенным реципиентам, у животных развилось миелопролиферативное заболевание, сильно напоминающее ХМЛ человека [117]. С тех пор эти модели широко использовались для оценки многих аспектов ХМЛ, включая трансформирующий потенциал различных изоформ BCR-ABL [118], генетику прогрессирования заболевания [36] и характеристику стволовых клеток ХМЛ [119]. .
CML также моделировали с использованием трансгенных мышей. В одной модели изоформа p210 экспрессировалась из индуцируемого тетрациклином протомера и скрещивалась с трансгенной мышью, которая экспрессировала tet-трансактиватор с промотора CD34, обеспечивая экспрессию p210 в присутствии доксициклина в компартменте HSC. В результате у мышей развилось миелопролиферативное заболевание с тромбоцитозом [120]. Это противоположно B-клеточному ALL, которое они наблюдали, когда p210 экспрессировался в пре-B-клетках [121], демонстрируя, что фенотип лейкозов, индуцированных BCR-ABL, зависит, по крайней мере, частично от клетки-источника.В дополнение к мозаичным и трансгенным моделям были проведены исследования ксенотрансплантатов с использованием образцов пациентов с ХМЛ. В недавнем примере сохранение популяции стволовых клеток ХМЛ у пациентов в цитогенетической ремиссии было продемонстрировано путем приживления клеток мышам NSG [122]. Это исследование доказывает устойчивость этих лейкозных стволовых клеток даже у пациентов, которые получали терапию TKI в течение многих лет. Первичные образцы пациентов и клеточные линии, полученные от пациентов с ХМЛ в различных фазах, были приживлены бестимусным мышам [123], мышам SCID [124] и мышам NOD / SCID [125].Использование мозаичных, трансгенных и ксенотрансплантных моделей дало неоценимую информацию о молекулярной патологии этого лейкоза.
Животные модели ХЛЛ
ХЛЛ является наиболее распространенной из всех лейкозов и, в отличие от ХМЛ, является генетически гетерогенным заболеванием. Это заболевание пожилых людей, характеризующееся клональной пролиферацией иммунодефицитных CD5 + зрелых В-лимфоцитов. Заболевание считается вялотекущим, при этом средняя выживаемость при низкой стадии заболевания составляет более десяти лет, хотя у подгруппы пациентов будет более скоротечное течение [126].Заболевание может иногда трансформироваться в более агрессивную диффузную крупноклеточную В-клеточную лимфому или, реже, в лимфому Ходжкина посредством процесса, называемого трансформацией Рихтера. К настоящему времени описано несколько моделей ХЛЛ на животных. Как и в случае с другими лейкозами, мыши являются наиболее широко используемой моделью.
Одной из самых ранних моделей мышей с ХЛЛ является спонтанно возникшая модель мышей новозеландских черных (NZB). У мышей NZB развивается клональная пролиферация анеуплоидных лимфоцитов CD5 + B. Заболевание может быть пересажено серийно и иногда превращается в крупноклеточную лимфому, соответствующую трансформации Рихтера, наблюдаемой клинически [127].У этих мышей была обнаружена мутация в локусе микроРНК-16; также наблюдаемый в клетках ХЛЛ человека, считается, что он способствует развитию ХЛЛ [128]. В дополнение к этой спонтанной модели было разработано несколько трансгенных моделей. В трансгенной модели TCL1 человеческий ген TCL1 был клонирован ниже промотора E, что привело к ограничению экспрессии B-клеток на высоком уровне. Ген TCL1 имеет повышенную экспрессию при ряде злокачественных новообразований В-клеток, включая ХЛЛ [129].Полученные мыши развивают клональную или олигоклональную пролиферацию лимфоцитов CD5 + B в более позднем возрасте, имитируя болезнь человека [130]. Дополнительное понимание молекулярной патологии ХЛЛ было получено с помощью двойной трансгенной модели TRAF2DN / Bcl-2. Как Bcl-2, так и факторы, ассоциированные с рецептором фактора некроза опухоли (TRAF), сверхэкспрессируются при злокачественных новообразованиях B-клеток, включая CLL [131–132]. В этой модели исследователи скрестили трансгенных животных, экспрессирующих человеческий BCL2 с промотора тяжелой цепи IgG, с животными, экспрессирующими усеченную форму TRAF2.Полученное потомство развивало прогрессивное накопление клональных CD-экспрессирующих В-клеток, что приводило к сокращению выживаемости [133]. Как и в случае с другими лейкозами, сообщалось о распространении образцов ХЛЛ человека у мышей с ослабленным иммунитетом. Полученная от человека линия клеток CLL, устойчивых к флударабину, WSUCLL, может вызывать опухоли при подкожном введении мышам SCID [134]. Эта модель использовалась для доклинических испытаний [135]. Голые мыши также служили реципиентами клеточных линий CLL человека [136]. Наконец, животных NOD / SCID использовали для приживления первичных образцов пациентов [137].Эта модель была использована для изучения эффектов известных прогностических маркеров ХЛЛ на приживление образца; образцы с неблагоприятными факторами прививаются более агрессивно в этой модели [138]. Первые успехи этой модели делают ее очень привлекательной для будущих доклинических исследований эффективности новых агентов.
Резюме и направления на будущее
Животные модели лейкемии были незаменимы в изучении этих разрушительных болезней. Можно привести убедительный аргумент в пользу того, что наше понимание лейкозов больше, чем любое другое злокачественное новообразование, выиграло от этих модельных систем.Большая часть нынешнего понимания и терапевтических подходов были впервые проверены или открыты с использованием этих моделей. В будущем можно легко представить синергию между различными моделями для дальнейшей разработки новых терапевтических целей. Трансгенные и мозаичные модели имеют очевидную полезность для доклинических испытаний эффективности, а также для изучения взаимодействий стромы и иммунных клеток. У моделей ксенотрансплантатов есть уникальное преимущество, заключающееся в том, что они могут исследовать специфическую биологию клеток человека в in vivo, хотя и в среде с ослабленным иммунитетом.Новые системы, не относящиеся к млекопитающим, могут привести к крупномасштабному генетическому скринингу для выявления критических молекулярных путей, которые могут служить следующим поколением терапевтических мишеней, которые будут оценены на мозаичных, трансгенных и ксенотрансплантных моделях и переведены в клинические испытания для улучшения жизни. наших пациентов с лейкемиями.
Благодарности
Авторы выражают благодарность Карен Кляйн за помощь в редактировании рукописи. Поддержка предоставлена Фондом Дуга Коли по исследованиям лейкемии, Фондом Маккея по исследованиям рака, Фондом исследований Лейта и Фондом Фрэнсис П. Тутуилер.
Библиография
1. Сигел Р., Уорд Э, Броули О., Джемаль А. Статистика рака, 2011: Влияние устранения социально-экономических и расовых различий на преждевременную смерть от рака. CA Cancer J Clin. 2011. 61: 212–236. [PubMed] [Google Scholar] 2. Гиллиланд Д.Г., Джордан Коннектикут, Феликс Калифорния. Молекулярные основы лейкемии. Образовательная программа Hematology Am Soc Hematol. 2004: 80–97. [PubMed] [Google Scholar] 3. Licht JD, Sternberg DW. Молекулярная патология острого миелолейкоза. Образовательная программа Hematology Am Soc Hematol.2005: 137–142. [PubMed] [Google Scholar] 4. Ловенберг Б. Острый миелоидный лейкоз: проблема выявления разнообразия болезней. Образовательная программа Hematology Am Soc Hematol. 2008; 2008: 1–11. [PubMed] [Google Scholar] 5. Dohner H, Estey EH, Amadori S и др. Диагностика и лечение острого миелоидного лейкоза у взрослых: рекомендации международной группы экспертов от имени European Leukemia Net. Кровь. 2010. 115: 453–474. [PubMed] [Google Scholar] 6. Нерви Б., Рамирес П., Реттиг М.П. и др. Хемосенсибилизация острого миелоидного лейкоза (ОМЛ) после мобилизации антагонистом CXCR4 AMD3100.Кровь. 2009. 113: 6206–6214. [Бесплатная статья PMC] [PubMed] [Google Scholar] 7. Jaiswal S, Jamieson CH, Pang WW, et al. CD47 активируется циркулирующими гемопоэтическими стволовыми клетками и лейкозными клетками, чтобы избежать фагоцитоза. Клетка. 2009. 138: 271–285. [Бесплатная статья PMC] [PubMed] [Google Scholar] 8. Маджети Р., Чао М.П., Ализаде А.А. и др. CD47 является неблагоприятным прогностическим фактором и мишенью терапевтических антител для стволовых клеток острого миелоидного лейкоза человека. Клетка. 2009. 138: 286–299. [Бесплатная статья PMC] [PubMed] [Google Scholar] 9.Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Лейкозные клетки создают ниши костного мозга, которые нарушают поведение нормальных кроветворных клеток-предшественников. Наука. 2008; 322: 1861–1865. [PubMed] [Google Scholar] 10. Лапидот Т., Сирард С., Формор Дж. И др. Клетка, инициирующая острый миелоидный лейкоз человека после трансплантации мышам SCID. Природа. 1994; 367: 645–648. [PubMed] [Google Scholar] 11. Джордан CT, Гусман ML, Noble M. Раковые стволовые клетки. N Engl J Med. 2006; 355: 1253–1261. [PubMed] [Google Scholar] 12.Skipper HE, Perry S. Кинетика нормальных и лейкозных популяций лейкоцитов и отношение к химиотерапии. Cancer Res. 1970; 30: 1883–1897. [PubMed] [Google Scholar] 13. Закон LW, Таормина V, Бойл PJ. Ответ острых лимфолейкозов на антагонист пуринов 6-меркаптопурин. Ann N Y Acad Sci. 1954. 60: 244–250. [PubMed] [Google Scholar] 14. Маккормак Э., Брюзруд О., Гьертсен БТ. Животные модели острого миелогенного лейкоза — разработка, применение и перспективы на будущее. Лейкемия. 2005; 19: 687–706.[PubMed] [Google Scholar] 15. Шкипер HE, Schabel FM, Jr, Wilcox WS. Экспериментальная оценка потенциальных противораковых агентов. XXI. Планирование приема арабинозилцитозина для использования его специфичности в S-фазе против лейкозных клеток. Cancer Chemother Rep. 1967; 51: 125–165. [PubMed] [Google Scholar] 16. Френд С. Бесклеточная передача у взрослых швейцарских мышей болезни, имеющей характер лейкемии. J Exp Med. 1957; 105: 307–318. [Бесплатная статья PMC] [PubMed] [Google Scholar] 17. Линемейер Д.Л., Менке Дж. Г., Рускетти С. К., Эванс Л. Х., Сколник Е. М..Последовательности гена оболочки, которые кодируют белок gp52 фокусообразующего вируса селезенки, необходимы для индукции пролиферации эритроидных клеток. J Virol. 1982; 43: 223–233. [Бесплатная статья PMC] [PubMed] [Google Scholar] 18. Вольф Л., Рускетти С. Злокачественная трансформация эритроидных клеток in vivo путем введения нереплицирующегося ретровирусного вектора. Наука. 1985; 228: 1549–1552. [PubMed] [Google Scholar] 19. Back J, Dierich A, Bronn C, Kastner P, Chan S. PU.1 определяет способность эритроидных клеток-предшественников к самообновлению.Кровь. 2004. 103: 3615–3623. [PubMed] [Google Scholar] 20. Erkeland SJ, Valkhof M, Heijmans-Antonissen C, et al. Масштабная идентификация генов болезней, вызывающих острый миелоидный лейкоз. J Virol. 2004; 78: 1971–1980. [Бесплатная статья PMC] [PubMed] [Google Scholar] 21. Кауделл Д., Харпер Д.П., Новак Р.Л. и др. Ретровирусный инсерционный мутагенез идентифицирует активацию Zeb2 как новое лейкемогенное событие взаимодействия у трансгенных мышей CALM-AF10. Кровь. 2010; 115: 1194–1203. [Бесплатная статья PMC] [PubMed] [Google Scholar] 22.Slape C, Hartung H, Lin YW, Bies J, Wolff L, Aplan PD. Ретровирусный инсерционный мутагенез идентифицирует гены, которые взаимодействуют с NUP98-HOXD13 во время лейкемической трансформации. Cancer Res. 2007. 67: 5148–5155. [Бесплатная статья PMC] [PubMed] [Google Scholar] 23. Шкипер HE, Schabel FM, младший, трейдер MW, Laster WR., Младший Ответ на терапию спонтанного, первого прохождения и длинных линий прохождения лейкемии AK. Cancer Chemother Rep., 1969; 53: 345–366. [PubMed] [Google Scholar] 24. Василиу Г.С., Купер Дж. Л., Рад Р. и др.Мутантный нуклеофозмин и взаимодействующие пути приводят к возникновению и прогрессированию лейкемии у мышей. Нат Жене. 2011; 43: 470–475. [Бесплатная статья PMC] [PubMed] [Google Scholar] 25. Коллиер Л.С., Адамс Д.Д., Хакетт С.С. и др. Мутагенез спящей красавицы всего тела может вызвать пенетрантную лейкемию / лимфому и редкую глиому высокой степени злокачественности без связанной с этим эмбриональной летальности. Cancer Res. 2009; 69: 8429–8437. [Бесплатная статья PMC] [PubMed] [Google Scholar] 26. Heyer J, Kwong LN, Lowe SW, Chin L. Генно-инженерные модели мышей, не являющиеся зародышевыми линиями, для трансляционных исследований рака.Нат Рев Рак. 2010; 10: 470–480. [Бесплатная статья PMC] [PubMed] [Google Scholar] 27. Early E, Moore MA, Kakizuka A, et al. Трансгенная экспрессия PML / RARalpha нарушает миелопоэз. Proc Natl Acad Sci U S. A. 1996; 93: 7900–7904. [Бесплатная статья PMC] [PubMed] [Google Scholar] 28. Браун Д., Коган С., Лагассе Е. и др. Трансген PMLRARalpha вызывает острый промиелоцитарный лейкоз у мышей. Proc Natl Acad Sci U S. A. 1997; 94: 2551–2556. [Бесплатная статья PMC] [PubMed] [Google Scholar] 29. Гризолано Дж. Л., Весселшмидт Р. Л., Пеличчи П. Г., Лей Т. Дж..Измененное миелоидное развитие и острый лейкоз у трансгенных мышей, экспрессирующих PML-RAR альфа под контролем регуляторных последовательностей катепсина G. Кровь. 1997. 89: 376–387. [PubMed] [Google Scholar] 30. He LZ, Guidez F, Tribioli C и др. Различное взаимодействие PML-RARalpha и PLZF-RARalpha с корепрессорами определяет дифференциальные ответы на RA в APL. Нат Жене. 1998. 18: 126–135. [PubMed] [Google Scholar] 31. Зубер Дж., Радтке И., Парди Т.С. и др. Мышиные модели ОМЛ человека точно предсказывают ответ на химиотерапию.Genes Dev. 2009; 23: 877–889. [Бесплатная статья PMC] [PubMed] [Google Scholar] 32. Гримуэйд Д., Уокер Х., Оливер Ф. и др. Важность диагностической цитогенетики для исходов при ОМЛ: анализ 1612 пациентов, включенных в исследование MRC AML-10. Рабочие группы Совета медицинских исследований по лейкемии взрослых и детей. Кровь. 1998. 92: 2322–2333. [PubMed] [Google Scholar] 33. Окуда Т., Цай З., Ян С. и др. Экспрессия заблокированного гена лейкемии AML1-ETO ингибирует установление нормального дефинитивного гематопоэза и непосредственно генерирует диспластические гематопоэтические предшественники.Кровь. 1998. 91: 3134–3143. [PubMed] [Google Scholar] 34. Роадс К.Л., Хетерингтон С.Дж., Харакава Н. и др. Анализ роли AML1-ETO в лейкемогенезе с использованием модели индуцибельных трансгенных мышей. Кровь. 2000; 96: 2108–2115. [PubMed] [Google Scholar] 35. Юань Ю., Чжоу Л., Миямото Т. и др. Экспрессия AML1-ETO напрямую участвует в развитии острого миелоидного лейкоза при наличии дополнительных мутаций. Proc Natl Acad Sci U S. A. 2001; 98: 10398–10403. [Бесплатная статья PMC] [PubMed] [Google Scholar] 36.Даш А.Б., Уильямс И.Р., Куток Дж. Л. и др. Мышиная модель бластного кризиса ХМЛ, вызванного сотрудничеством между BCR / ABL и NUP98 / HOXA9. Proc Natl Acad Sci U S. A. 2002; 99: 7622–7627. [Бесплатная статья PMC] [PubMed] [Google Scholar] 37. Гризолано Дж. Л., О’Нил Дж., Каин Дж., Томассон М. Х. Активированная рецепторная тирозинкиназа, TEL / PDGFbetaR, взаимодействует с AML1 / ETO, вызывая острый миелоидный лейкоз у мышей. Proc Natl Acad Sci U S. A. 2003; 100: 9506–9511. [Бесплатная статья PMC] [PubMed] [Google Scholar] 38. Lavau C, Du C, Thirman M, Zeleznik-Le N.Связанные с хроматином свойства CBP, слитого с MLL, вызывают миелодиспластический синдром, который перерастает в миелоидный лейкоз. EMBO J. 2000; 19: 4655–4664. [Бесплатная статья PMC] [PubMed] [Google Scholar] 39. Смотреть на. Онкогенные факторы транскрипции при острых лейкозах человека. Наука. 1997; 278: 1059–1064. [PubMed] [Google Scholar] 40. Daser A, Rabbitts TH. Расширение репертуара гена лейкемии смешанного происхождения MLL в лейкемогенезе. Genes Dev. 2004; 18: 965–974. [PubMed] [Google Scholar] 41. Corral J, Lavenir I, Impey H и др.Ген слияния Mll-AF9, полученный путем гомологичной рекомбинации, вызывает острый лейкоз у химерных мышей: метод создания слитых онкогенов. Клетка. 1996. 85: 853–861. [PubMed] [Google Scholar] 42. Стриссель П.Л., Стрик Р., Томек Р.Дж., Роу Б.А., Роули Д.Д., Железник-Ле, штат Нью-Джерси. Структурные свойства ДНК AF9 аналогичны MLL и могут действовать как горячие точки рекомбинации, приводящие к транслокациям MLL / AF9 и лейкемогенезу. Hum Mol Genet. 2000; 9: 1671–1679. [PubMed] [Google Scholar] 43. Коллинз Э.С., Паннелл Р., Симпсон Э.М., Форстер А., Кролик Т.Х.Межхромосомная рекомбинация генов Mll и Af9, опосредованная cre-loxP в развитии мышей. EMBO Rep. 2000; 1: 127–132. [Бесплатная статья PMC] [PubMed] [Google Scholar] 44. Форстер А., Паннелл Р., Дринан Л. Ф. и др. Разработка de novo реципрокных хромосомных транслокаций, связанных с Mll, для репликации первичных событий рака человека. Раковая клетка. 2003. 3: 449–458. [PubMed] [Google Scholar] 45. Стируолт Д.Л., Радич Ю.П. Роль FLT3 в злокачественных заболеваниях кроветворения. Нат Рев Рак. 2003. 3: 650–665. [PubMed] [Google Scholar] 46.Ли Л., Пилото О., Нгуен Х. Б. и др. Включение внутренней тандемной дупликационной мутации в мышиный FLT3 вызывает миелопролиферативное заболевание на мышиной модели. Кровь. 2008; 111: 3849–3858. [Бесплатная статья PMC] [PubMed] [Google Scholar] 47. Гринблатт С., Ли Л., Слейп С и др. Включение мутации FLT3 / ITD кооперируется со слиянием NUP98-HOXD13 для генерации острого миелоидного лейкоза на модели мышей. Кровь. 2012 [Бесплатная статья PMC] [PubMed] [Google Scholar] 48. Чан ИТ, Гиллиланд Д.Г. Онкогенные K-ras в мышиных моделях миелопролиферативного заболевания и острого миелоидного лейкоза.Клеточный цикл. 2004; 3: 536–537. [PubMed] [Google Scholar] 49. Маккензи К.Л., Дольников А., Миллингтон М., Шоунан Ю., Симондс Г. Мутантные N-расы вызывают миелопролиферативные нарушения и апоптоз у мышей с репопуляцией костного мозга. Кровь. 1999; 93: 2043–2056. [PubMed] [Google Scholar] 50. Мартелли AM, Nyakern M, Tabellini G, et al. Сигнальный путь фосфоинозитид-3-киназы / Akt и его терапевтические последствия для острого миелоидного лейкоза человека. Лейкемия. 2006; 20: 911–928. [PubMed] [Google Scholar] 51. Ю Х, Ли Й, Гао С. и др.Соответствующая мышиная модель моноцитарного лейкоза человека посредством Cre / lox-контролируемой миелоид-специфической делеции PTEN. Лейкемия. 2010; 24: 1077–1080. [Бесплатная статья PMC] [PubMed] [Google Scholar] 52. Каттс Б.А., Шегрен А.К., Андерссон К.М. и др. Дефицит Nf1 взаимодействует с онкогенным K-RAS, вызывая у мышей острый миелоидный лейкоз. Кровь. 2009. 114: 3629–3632. [PubMed] [Google Scholar] 53. de Guzman CG, Warren AJ, Zhang Z, et al. Экспансия гемопоэтических стволовых клеток и отчетливые аномалии развития миелоидов на мышиной модели транслокации AML1-ETO.Mol Cell Biol. 2002; 22: 5506–5517. [Бесплатная статья PMC] [PubMed] [Google Scholar] 54. Парди Т.С., Зубер Дж., Лоу SW. Flt3-ITD изменяет химиотерапевтический ответ in vitro и in vivo p53-зависимым образом. Exp Hematol. 2011; 39: 473–485. e474. [Бесплатная статья PMC] [PubMed] [Google Scholar] 55. Bruserud O, Tore Gjertsen B, Brustugun OT и др. Влияние интерлейкина 10 на бластные клетки, полученные от пациентов с острым миелогенным лейкозом. Лейкемия. 1995; 9: 1910–1920. [PubMed] [Google Scholar] 56. Bruserud O, Gjertsen BT, von Volkman HL.Культура in vitro клеток острого миелогенного лейкоза человека (AML) в бессывороточной среде: исследования нативных бластов AML и клеточных линий AML. J Hematother Stem Cell Res. 2000; 9: 923–932. [PubMed] [Google Scholar] 57. Нара Н., Миямото Т. Прямая и серийная трансплантация острого миелоидного лейкоза человека голым мышам. Br J Рак. 1982; 45: 778–782. [Бесплатная статья PMC] [PubMed] [Google Scholar] 58. Sawyers CL, Gishizky ML, Quan S, Golde DW, Witte ON. Распространение бластных миелоидных лейкозов человека у мышей SCID.Кровь. 1992; 79: 2089–2098. [PubMed] [Google Scholar] 59. Ailles LE, Gerhard B, Kawagoe H, Hogge DE. Характеристики роста предшественников острого миелогенного лейкоза, которые инициируют злокачественное кроветворение у мышей, не страдающих ожирением, диабетом / тяжелым комбинированным иммунодефицитом. Кровь. 1999; 94: 1761–1772. [PubMed] [Google Scholar] 60. Вундерлих М., Чоу Ф.С., Линк К.А. и др. Эффективность ксенотрансплантата AML значительно повышается у мышей NOD / SCID-IL2RG, конститутивно экспрессирующих SCF, GM-CSF и IL-3 человека. Лейкемия. 2010; 24: 1785–1788.[Бесплатная статья PMC] [PubMed] [Google Scholar] 61. Svejda J, Kossey P, Hlavayova E, Svec F. Гистологическая картина лейкемии трансплантируемых крыс, вызванная рентгеновским облучением и метилхолантреном. Новообразования. 1958; 5: 123–131. [PubMed] [Google Scholar] 62. Мориучи Т., Оикава Т., Кодама Т., Ямагути Х., Кобаяси Х. Создание и характеристика трансплантируемого миеломоноцитарного лейкоза крыс. Cancer Res. 1983; 43: 5478–5483. [PubMed] [Google Scholar] 63. Иванкович С., Целлер В.Дж. [Лейкемия L 5222 штамма крыс BD IX.Индуцированная этилнитрозомочевиной моноцитарно-миелоидная трансплантируемая форма для цитохимических и химиотерапевтических исследований] Блют. 1974. 28: 288–292. [PubMed] [Google Scholar] 64. Пирсон Дж. У., Чапарас С. Д., Торгерсен Дж. А., Перк К., Чиригос М. А., Шер Н. А.. Эффект лекарственной терапии против гистологически определенного лейкоза крыс. Cancer Res. 1974; 34: 355–361. [PubMed] [Google Scholar] 65. Zeller WJ, Ivankovic S, Schmahl D. Химиотерапия трансплантируемого острого лейкоза L5222 у крыс. Cancer Res. 1975. 35: 1168–1174. [PubMed] [Google Scholar] 66.Hagenbeek A, Martens AC. Эффективность высоких доз циклофосфамида в сочетании с облучением всего тела при лечении острого миелоцитарного лейкоза: исследования на соответствующей модели крыс. Cancer Res. 1983; 43: 408–412. [PubMed] [Google Scholar] 67. Hagenbeek A, Martens AC. Патогенез крысиной модели острого миелоцитарного лейкоза человека. Haematologica. 1980; 65: 293–308. [PubMed] [Google Scholar] 68. ван Беккум Д.В., ван Остером П., Дике К.А. Образование колоний in vitro при трансплантируемых лейкозах крыс по сравнению с острым миелоидным лейкозом человека.Cancer Res. 1976; 36: 941–946. [PubMed] [Google Scholar] 69. Martens AC, van Bekkum DW, Hagenbeek A. Минимальная остаточная болезнь при лейкемии: исследования на животной модели острого миелоцитарного лейкоза (BNML) Int J Cell Cloning. 1990; 8: 27–38. [PubMed] [Google Scholar] 70. Пруво Б., Жакель А., Дроин Н. и др. Ксенотрансплантат лейкозных клеток в эмбрионе рыбок данио для исследования эффективности лекарств. Haematologica. 2011; 96: 612–616. [Бесплатная статья PMC] [PubMed] [Google Scholar] 71. Коркери Д.П., Деллер Г., Берман Дж. Ксенотрансплантация лейкемии в анализе реакции на химиотерапию рыбок данио in vivo.Br J Haematol. 2011; 153: 786–789. [PubMed] [Google Scholar] 72. Osman D, Gobert V, Ponthan F, Heidenreich O, Haenlin M, Waltzer L. Модель дрозофилы идентифицирует кальпаины как модуляторы человеческого лейкемогенного слитого белка AML1-ETO. Proc Natl Acad Sci U S. A. 2009; 106: 12043–12048. [Бесплатная статья PMC] [PubMed] [Google Scholar] 73. Hunger SP, Lu X, Devidas M и др. Улучшение выживаемости детей и подростков с острым лимфобластным лейкозом в период с 1990 по 2005 год: отчет группы детской онкологии.J Clin Oncol. 2012 [Бесплатная статья PMC] [PubMed] [Google Scholar] 74. Груша В.С., Астер Дж. К., Скотт М.Л. и др. Исключительное развитие Т-клеточных новообразований у мышей с трансплантированным костным мозгом, экспрессирующим активированные аллели Notch. J Exp Med. 1996; 183: 2283–2291. [Бесплатная статья PMC] [PubMed] [Google Scholar] 75. Heisterkamp N, Jenster G, ten Hoeve J, Zovich D, Pattengale PK, Groffen J. Острый лейкоз у трансгенных мышей bcr / abl. Природа. 1990; 344: 251–253. [PubMed] [Google Scholar] 76. Law LW, Dunn TB и др. Наблюдения за действием антагониста фолиевой кислоты на трансплантируемые лимфоидные лейкозы у мышей.J Natl Cancer Inst. 1949; 10: 179–192. [PubMed] [Google Scholar] 77. Тренер DL, Уилок Э.Ф. Характеристика фенотипов клеток L5178Y, выделенных во время прогрессирования состояния покоя опухоли у мышей DBA2. Cancer Res. 1984; 44: 2897–2906. [PubMed] [Google Scholar] 78. Нишимура Т., Муто К., Танака Н. Лекарственная чувствительность устойчивой к адриамицину мутантной сублинии клеток лимфобластомы мыши L5178Y. Дж. Антибиотик (Токио) 1978; 31: 493–495. [PubMed] [Google Scholar] 79. Нисимура Т., Сузуки Х., Муто К., Танака Н. Механизм устойчивости к адриамицину в подлинии клеток лимфобластомы мыши L5178Y.Журнал «Антибиотик» (Токио), 1979; 32: 518–522. [PubMed] [Google Scholar] 80. Клойд М.В., Хартли Дж. В., Роу В.П. Лимфомагенность рекомбинантных вирусов мышиного лейкоза, индуцирующих очаговые клетки норки. J Exp Med. 1980; 151: 542–552. [Бесплатная статья PMC] [PubMed] [Google Scholar] 81. Юн Дж. П., Бехан Дж. В., Хейстеркамп Н. и др. Ожирение, вызванное диетой, ускоряет прогрессирование острого лимфобластного лейкоза в двух моделях на мышах. Cancer Prev Res (Phila) 2010; 3: 1259–1264. [Бесплатная статья PMC] [PubMed] [Google Scholar] 82. Weiser KC, Liu B, Hansen GM и др.Ретровирусные вставки в базе данных VISION идентифицируют молекулярные пути лимфолейкоза и лимфомы мышей. Мамм Геном. 2007. 18: 709–722. [Бесплатная статья PMC] [PubMed] [Google Scholar] 83. Деттман Э.Дж., Симко С.Дж., Аянга Б. и др. Prdm14 инициирует лимфобластный лейкоз после увеличения популяции клеток, напоминающих обычные лимфоидные предшественники. Онкоген. 2011; 30: 2859–2873. [Бесплатная статья PMC] [PubMed] [Google Scholar] 84. Groffen J, Voncken JW, Kaartinen V, Morris C, Heisterkamp N. Ph-позитивный лейкоз: модель трансгенных мышей.Лимфома лейка. 1993; 11 (Приложение 1): 19–24. [PubMed] [Google Scholar] 85. Reichert A, Heisterkamp N, Daley GQ, Groffen J. Лечение Bcr / Abl-положительного острого лимфобластного лейкоза у трансгенных мышей P190 с помощью ингибитора фарнезилтрансферазы SCH66336. Кровь. 2001; 97: 1399–1403. [PubMed] [Google Scholar] 86. Каур П., Фельдхан Н., Чжан Б. и др. Лечение нилотинибом на мышиных моделях лимфобластного лейкоза P190 Bcr / Abl. Молочный рак. 2007; 6: 67. [Бесплатная статья PMC] [PubMed] [Google Scholar] 87. Чен В., Ли К., Хадсон В. А., Кумар А., Кирххоф Н., Керси Дж. Х.Модель «нокаута» на мышах Mll-AF4 приводит к нарушению регуляции лимфоидной и миелоидной регуляции и гематологическому злокачественному новообразованию. Кровь. 2006. 108: 669–677. [Бесплатная статья PMC] [PubMed] [Google Scholar] 88. Мецлер М., Форстер А., Паннелл Р. и др. Условная модель опухолевого генеза B-клеток MLL-AF4 с использованием инверторной технологии. Онкоген. 2006; 25: 3093–3103. [PubMed] [Google Scholar] 89. Тамай Х., Мияке К., Такатори М. и др. Активированный белок K-Ras ускоряет индуцированную MLL / AF4 лейкемолимфомогенность человека в модели трансгенных мышей.Лейкемия. 2011; 25: 888–891. [PubMed] [Google Scholar] 90. Харрис А. В., Пинкерт Калифорния, Кроуфорд М., Лэнгдон В. Ю., Бринстер Р. Л., Адамс Дж. М.. Трансгенная мышь E. mu-myc. Модель спонтанной лимфомы с высокой заболеваемостью и лейкемии ранних В-клеток. J Exp Med. 1988. 167: 353–371. [Бесплатная статья PMC] [PubMed] [Google Scholar] 91. Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW. Мутации INK4a / ARF ускоряют лимфомагенез и повышают химиорезистентность, отключая p53. Genes Dev. 1999; 13: 2670–2677.[Бесплатная статья PMC] [PubMed] [Google Scholar] 92. Шмитт К.А., Фридман Дж. С., Янг М. и др. Программа старения, контролируемая p53 и p16INK4a, способствует результату лечения рака. Клетка. 2002. 109: 335–346. [PubMed] [Google Scholar] 93. Grabher C, von Boehmer H, Look AT. Активация Notch 1 в молекулярном патогенезе Т-клеточного острого лимфобластного лейкоза. Нат Рев Рак. 2006; 6: 347–359. [PubMed] [Google Scholar] 94. Эллисен Л.В., Берд Дж., Западный округ Колумбия и др. TAN-1, человеческий гомолог гена notch дрозофилы, разрушается хромосомными транслокациями в Т-лимфобластных новообразованиях.Клетка. 1991; 66: 649–661. [PubMed] [Google Scholar] 95. Вен А. П., Феррандо А. А., Ли В. и др. Активирующие мутации NOTCh2 при остром лимфобластном лейкозе Т-клеток человека. Наука. 2004. 306: 269–271. [PubMed] [Google Scholar] 96. Deftos ML, Huang E, Ojala EW, Forbush KA, Bevan MJ. Передача сигналов Notch2 способствует созреванию тимоцитов CD4 и CD8 SP. Иммунитет. 2000. 13: 73–84. [Бесплатная статья PMC] [PubMed] [Google Scholar] 97. Фаулкс Б.Дж., Роби Е.А. Переоценка влияния активированного Notch2 на развитие CD4 и CD8 Т-клеток.J Immunol. 2002; 169: 1817–1821. [PubMed] [Google Scholar] 98. Priceputu E, Bouallaga I, Zhang Y, et al. Структурно различные лиганд-связывающие или лиганд-независимые мутанты Notch2 являются лейкемогенными, но по-разному влияют на развитие тимоцитов, апоптоз и метастазирование. J Immunol. 2006; 177: 2153–2166. [PubMed] [Google Scholar] 99. Berquam-Vrieze KE, Swing DA, Tessarollo L, Dupuy AJ. Характеристика трансгенных мышей, экспрессирующих ассоциированные с раком варианты человеческого NOTCh2. Бытие. 2012; 50: 112–118. [Бесплатная статья PMC] [PubMed] [Google Scholar] 100.Boulos N, Mulder HL, Calabrese CR, et al. Химиотерапевтические агенты предотвращают появление устойчивых к дазатинибу мутаций киназы BCR-ABL на точной мышиной модели острого лимфобластного лейкоза с положительной хромосомой в Филадельфии. Кровь. 2011. 117: 3585–3595. [Бесплатная статья PMC] [PubMed] [Google Scholar] 101. Duy C, Hurtz C, Shojaee S и др. BCL6 позволяет Ph + клеткам острого лимфобластного лейкоза выжить при ингибировании киназы BCR-ABL1. Природа. 2011; 473: 384–388. [Бесплатная статья PMC] [PubMed] [Google Scholar] 102.Ван П.Й., Янг Ф., Чен С.Ю. и др. Биологические свойства лейкозов, возникающих в результате BCR / ABL-опосредованной трансформации, варьируются в зависимости от онтогенетического происхождения и активности гена p19ARF. Кровь. 2008; 112: 4184–4192. [Бесплатная статья PMC] [PubMed] [Google Scholar] 103. Барабе Ф., Кеннеди Дж. А., Хоуп К. Дж., Дик Дж. Э. Моделирование возникновения и прогрессирования острого лейкоза человека у мышей. Наука. 2007; 316: 600–604. [PubMed] [Google Scholar] 104. Вэй Дж., Вундерлих М., Фокс С. и др. Микроокружение определяет судьбу клонов в человеческой модели лейкемии MLL-AF9.Раковая клетка. 2008. 13: 483–495. [Бесплатная статья PMC] [PubMed] [Google Scholar] 105. le Viseur C, Hotfilder M, Bomken S и др. При остром лимфобластном лейкозе у детей бласты на разных стадиях иммунофенотипического созревания обладают свойствами стволовых клеток. Раковая клетка. 2008; 14: 47–58. [Бесплатная статья PMC] [PubMed] [Google Scholar] 106. Чиу П.П., Цзян Х., Дик Дж. Э. Клетки, инициирующие лейкоз при Т-лимфобластном лейкозе человека, проявляют устойчивость к глюкокортикоидам. Кровь. 2010; 116: 5268–5279. [PubMed] [Google Scholar] 107.Отова Б., Сладка М., Панчак А., Маринов И. Биологические характеристики спонтанных трансплантируемых Т-клеточных лимфом у инбредных крыс Sprague-Dawley / детенышей. Transplant Proc. 1997; 29: 1754–1755. [PubMed] [Google Scholar] 108. Отова Б., Вацлавикова Р., Даниелова В. и др. Эффекты паклитаксела, доцетаксела и их комбинаций на подкожные лимфомы у инбредных крыс Sprague-Dawley / Cub. Eur J Pharm Sci. 2006. 29: 442–450. [PubMed] [Google Scholar] 110. Sabaawy HE, Azuma M, Embree LJ, Tsai HJ, Starost MF, Hickstein DD.TELAML1 трансгенная модель рыбок данио предшественника В-клеточного острого лимфобластного лейкоза. Proc Natl Acad Sci U S. A. 2006; 103: 15166–15171. [Бесплатная статья PMC] [PubMed] [Google Scholar] 111. Фрейзер Дж. К., Микер Н. Д., Руднер Л. и др. Модели наследственных Т-клеточных злокачественных новообразований, установленные при фенотипическом скрининге у рыбок данио. Лейкемия. 2009; 23: 1825–1835. [Бесплатная статья PMC] [PubMed] [Google Scholar] 112. Лангенау Д.М., Травер Д., Феррандо А.А. и др. Myc-индуцированный Т-клеточный лейкоз у трансгенных рыбок данио. Наука. 2003. 299: 887–890.[PubMed] [Google Scholar] 113. Feng H, Langenau DM, Madge JA, et al. Индукция теплового шока Т-клеточной лимфомы / лейкемии у условно регулируемых Cre / lox трансгенных рыбок данио. Br J Haematol. 2007. 138: 169–175. [PubMed] [Google Scholar] 114. Feng H, Stachura DL, White RM и др. Клетки Т-лимфобластной лимфомы экспрессируют высокие уровни BCL2, S1P1 и ICAM1, что приводит к блокаде интравазации опухолевых клеток. Раковая клетка. 2010. 18: 353–366. [Бесплатная статья PMC] [PubMed] [Google Scholar] 115. Nowell PC, Hungerford DA.Хромосомные исследования при лейкемии человека. IV. Миелопролиферативный синдром и другие атипичные миелоидные нарушения. J Natl Cancer Inst. 1962; 29: 911–931. [PubMed] [Google Scholar] 116. Goldman JM. Начальное лечение пациентов с ХМЛ. Образовательная программа Hematology Am Soc Hematol. 2009: 453–460. [PubMed] [Google Scholar] 117. Дейли GQ, Ван Эттен Р.А., Балтимор Д. Индукция хронического миелогенного лейкоза у мышей геном P210bcr / abl филадельфийской хромосомы. Наука. 1990; 247: 824–830. [PubMed] [Google Scholar] 118.Ли С., Илария Р.Л., младший, Миллион РП, Дейли Г.К., Ван Эттен Р.А. Формы P190, P210 и P230 онкогена BCR / ABL вызывают аналогичный хронический миелолейкозоподобный синдром у мышей, но обладают различной лимфоидной лейкемогенной активностью. J Exp Med. 1999; 189: 1399–1412. [Бесплатная статья PMC] [PubMed] [Google Scholar] 119. Zhang H, Li H, Xi HS, Li S. HIF1alpha необходим для поддержания выживания стволовых клеток хронического миелоидного лейкоза. Кровь. 2012; 119: 2595–2607. [Бесплатная статья PMC] [PubMed] [Google Scholar] 120. Huettner CS, Koschmieder S, Iwasaki H, et al.Индуцируемая экспрессия BCR / ABL с использованием регуляторных элементов CD34 человека приводит к мегакариоцитарному миелопролиферативному синдрому. Кровь. 2003. 102: 3363–3370. [PubMed] [Google Scholar] 121. Хюттнер К.С., Чжан П., Ван Эттен Р.А., Тенен Д.Г. Обратимость острого В-клеточного лейкоза, вызванного BCR-ABL1. Нат Жене. 2000; 24: 57–60. [PubMed] [Google Scholar] 122. Чу С., Макдональд Т., Лин А. и др. Сохранение стволовых клеток лейкемии у пациентов с хроническим миелолейкозом в длительной ремиссии при лечении иматинибом.Кровь. 2011. 118: 5565–5572. [Бесплатная статья PMC] [PubMed] [Google Scholar] 123. Lozzio BB, Lozzi CB, Machado E. Линия клеток миелогенного (Ph +) лейкоза человека: трансплантация бестимусным мышам. J Natl Cancer Inst. 1976; 56: 627–629. [PubMed] [Google Scholar] 124. Skorski T, Nieborowska-Skorska M, Nicolaides NC и др. Подавление роста лейкозных клеток Philadelphia1 у мышей антисмысловым олигодезоксинуклеотидом BCR-ABL. Proc Natl Acad Sci U S. A. 1994; 91: 4504–4508. [Бесплатная статья PMC] [PubMed] [Google Scholar] 125.Дацци Ф., Хассерджиан Р., Гордон М.Ю. и др. Гемопоэтические клетки ХМЛ нормальной и хронической фаз повторно заселяют костный мозг NOD / SCID с различной кинетикой и представлением клеточных клонов. Hematol J. 2000; 1: 307–315. [PubMed] [Google Scholar] 126. Dohner H, Stilgenbauer S, Benner A и др. Геномные аберрации и выживаемость при хроническом лимфолейкозе. N Engl J Med. 2000; 343: 1910–1916. [PubMed] [Google Scholar] 127. Филлипс Дж. А., Мехта К., Фернандес С., Равече Е. С.. Мышь NZB как модель хронического лимфолейкоза.Cancer Res. 1992; 52: 437–443. [PubMed] [Google Scholar] 128. Равече Э.С., Салерно Э., Скаглионе Б.Дж. и др. Аномальный локус микроРНК-16 с синтенией человеческого 13q14, связанный с ХЛЛ у мышей NZB. Кровь. 2007; 109: 5079–5086. [Бесплатная статья PMC] [PubMed] [Google Scholar] 129. Нардуччи М.Г., Пескармона Э., Лазцери С. и др. Регуляция экспрессии TCL1 в B- и T-клеточных лимфомах и реактивных лимфоидных тканях. Cancer Res. 2000; 60: 2095–2100. [PubMed] [Google Scholar] 130. Бичи Р., Шинтон С.А., Мартин Э.С. и др. Хронический лимфоцитарный лейкоз человека, моделируемый на мышах с помощью направленной экспрессии TCL1.Proc Natl Acad Sci U S. A. 2002; 99: 6955–6960. [Бесплатная статья PMC] [PubMed] [Google Scholar] 131. Агилар-Сантелизес М., Роттенберг М.Э., Левин Н., Меллштедт Н., Йондал М. Экспрессия Bcl-2, Bax и p53 в B-CLL в зависимости от выживаемости и клинического прогрессирования in vitro. Int J Cancer. 1996; 69: 114–119. [PubMed] [Google Scholar] 132. Munzert G, Kirchner D, Stobbe H, et al. Сверхэкспрессия гена фактора некроза опухоли, связанного с рецептором фактора 1, при В-клеточном хроническом лимфоцитарном лейкозе: анализ ингибиторов апоптоза, регулируемых NF-каппа B / Rel.Кровь. 2002; 100: 3749–3756. [PubMed] [Google Scholar] 133. Сапата Дж. М., Краевская М., Морс ХК, 3-й, Цой Й., Рид Дж. Домен TNF-рецепторассоциированного фактора (TRAF) и Bcl-2 взаимодействуют, вызывая малую В-клеточную лимфому / хронический лимфоцитарный лейкоз у трансгенных мышей. Proc Natl Acad Sci U S. A. 2004; 101: 16600–16605. [Бесплатная статья PMC] [PubMed] [Google Scholar] 134. Мохаммад Р.М., Мохамед А.Н., Хамдан М.Ю. и др. Создание модели ксенотрансплантата B-CLL человека: полезность в качестве доклинической терапевтической модели. Лейкемия.1996. 10: 130–137. [PubMed] [Google Scholar] 135. Мохаммад Р.М., Лимварапус С., Хамди Н. и др. Обработка модели ксенотрансплантата de novo устойчивой к флударабину ХЛЛ бриостатином 1 с последующим введением флударабина. Int J Oncol. 1999; 14: 945–950. [PubMed] [Google Scholar] 136. Loisel S, Ster KL, Quintin-Roue I, et al. Создание новой модели человеческого B-CLL-подобного ксенотрансплантата на мышах nude. Leuk Res. 2005; 29: 1347–1352. [PubMed] [Google Scholar] 137. Дуриг Дж., Эбелинг П., Грабеллус Ф. и др. Новая модель ксенотрансплантата, не связанного с ожирением, диабетом и тяжелым иммунодефицитом для хронического лимфолейкоза, отражает важные клинические характеристики заболевания.Cancer Res. 2007; 67: 8653–8661. [PubMed] [Google Scholar] 138. Aydin S, Grabellus F, Eisele L, et al. Изучение роли CD38 и функционально связанных молекулярных факторов риска в модели ксенотрансплантата CLL NOD / SCID. Eur J Haematol. 2011; 87: 10–19. [PubMed] [Google Scholar]Что-нибудь ближе к реальности?
Метастаз рака Ред. Авторская рукопись; доступно в PMC 2014 1 июня.
Опубликован в окончательной отредактированной форме как:
PMCID: PMC3568447
NIHMSID: NIHMS416371
Раздел по гематологии и онкологии, Wake Forest Baptist Health, Уинстон-Сейлем, другие статьи в ЧВК, цитирующих опубликованную статью.
Abstract
Модели на животных сыграли неоценимую роль в усилиях по лучшему пониманию и лечению пациентов, страдающих лейкемией. Несмотря на то, что на основе этих моделей были сделаны важные выводы, следует признать ограничения. В этом обзоре мы выделим различные модели лейкемии на животных и опишем их вклад в улучшение понимания и лечения этих видов рака.
Введение
Лейкемии представляют собой разнообразную группу заболеваний, которые приводят к значительной заболеваемости и смертности.В 2011 году у более 44 600 американцев была диагностирована лейкемия, что привело к 21 780 смертельным исходам [1]. Четыре основных типа составляют 85% всех лейкозов: острый миелоидный лейкоз (AML), острый лимфобластный лейкоз (ALL), хронический миелоидный лейкоз (CML) и хронический лимфолейкоз (CLL). Этим и будет посвящен этот обзор. ХЛЛ является наиболее распространенным заболеванием, но на ОМЛ приходится ~ 42% всех смертей от лейкемии [1]. В результате ОМЛ является наиболее интенсивно изучаемым лейкозом и имеет самую разнообразную группу моделей на животных.Трудно переоценить влияние животных моделей на исследования лейкемии. Начиная с новаторской работы доктора Ллойда Лоу о реакции мышиного лимфоидного лейкоза на антиметаболиты в 1940-х и 1950-х годах до открытия лейкемической стволовой клетки доктором Джоном Диком в 1990-х годах, животные модели были неотъемлемой частью нашего понимания биология лейкозов.
Модели ОМЛ на животных
Введение
За последние 40 лет наше понимание молекулярной патофизиологии ОМЛ значительно выросло (обзор в [2–4]).Заболевание проявляется увеличением незрелых, клональных, миелоидных предшественников, что приводит к прогрессирующей недостаточности костного мозга и, в конечном итоге, к смерти. Несмотря на лучшее понимание генетики ОМЛ, терапия для большинства пациентов осталась неизменной, а общая 5-летняя выживаемость составляет 30-40% [5]. Этот неутешительный показатель выживаемости вдохновил на большую работу по разработке более подходящих моделей на животных для использования при обнаружении новых целей и тестировании новых методов лечения. В результате несколько исследований выявили новые идеи, которые стали возможными только благодаря использованию моделей на животных.Несколько примечательных примеров включают роль взаимодействия клеток AML / стромы костного мозга в устойчивости к химиотерапии [6], способность иммунной системы взаимодействовать с AML и нацеливаться на них [7-8], манипулирование нишей гемопоэтических стволовых клеток посредством AML [9], а также плодотворное наблюдение, что AML существует как иерархия клеток с редкой популяцией лейкемических стволовых клеток [10]. Это наблюдение стало центром теории раковых стволовых клеток, которая теперь распространяется и на многие солидные опухоли (см. Обзор [11]).
Мышиные модели
С 1930-х гг. Мышиные модели наиболее широко использовались для изучения ОМЛ. Первоначальные трансплантируемые модели, индуцированные канцерогенами, уступили место более современным трансгенным, ксенотрансплантатным и мозаичным подходам. В этом разделе будут рассмотрены основные типы мышиных моделей AML.
Канцероген-индуцированные модели
Среди первых описанных моделей мышиного лейкоза были трансплантируемые канцерогенные модели, используемые для тестирования возможных терапевтических агентов (см.).Использование антиметаболитных агентов было впервые протестировано на этих моделях. У них были преимущества, заключающиеся в том, что они могли размножать in vitro и затем вводить их большим когортам реципиентов, у которых впоследствии разовьется лейкемия, что позволило провести испытания многообещающих агентов и графиков. Они были использованы с большим эффектом докторами Скиппером и Шабелем для описания кинетики лейкемогенеза и ответа на терапию, которые остаются основной опорой нашего современного понимания болезни (см. Обзор в [12]).Одной из наиболее широко используемых моделей является линия клеток L1210, выделенная доктором Лоу путем воздействия на мышей DBA / 2 канцерогенного 3-метилхолантрена (3-MC) [13]. Полученный лейкоз был получен от умирающего животного и десятилетиями использовался экспериментально. Дополнительные химически индуцированные линии клеток мышиного лейкоза включают, среди прочего, P388, P1534 и L5178Y (обзор см. В [14]). Однако у этих моделей есть несколько недостатков. Самым сильным из них является тот факт, что патогенез этого лейкоза не имеет отношения к большинству случаев ОМЛ у людей, поскольку у немногих людей лейкоз развивается после воздействия химических агентов.Действительно, по большей части считалось, что некоторые из этих клеточных линий более тесно связаны с лимфоидными злокачественными новообразованиями, чем ОМЛ. Кроме того, генетические основы болезни неизвестны, что ограничивает применимость результатов с использованием этих моделей. Несмотря на эти недостатки, эти модели внесли значительный вклад в лечение ОМЛ, особенно в поддержку использования цитарабина [15], который остается одним из наиболее широко используемых агентов для лечения ОМЛ и ОЛЛ.
Основные методы создания моделей лейкемии на животных.А) Модели, индуцированные канцерогеном. Животные из восприимчивых штаммов подвергаются воздействию канцерогенных химических веществ или ионизирующего излучения. После заражения животные наблюдаются на предмет развития лейкемии. Б) Мозаика, модели транспозонов и вирусов. В мозаичных моделях HSC собирают от мышей-доноров и заражают вирусами, кодирующими интересующий онкоген. Интеграция вируса в геном хозяина приводит к доставке и экспрессии онкогена. В дополнение к доставке онкогенов внедрение вируса может приводить к аберрантной экспрессии генов клеточных протоонкогенов (как показано) или к нарушению клеточного супрессора опухоли, если интеграция происходит внутригенно.Мутагенез транспозонов является результатом аналогичных последствий интеграции транспозонов в геном хозяина. В) Трансгенные модели. Направляющий вектор, который был сконструирован для экспрессии интересующего трансгена, инъецировали или электропорировали в мышиные ES-клетки. ES-клетки, которые интегрировали вектор, затем вводятся в тетраплоидные бластоцисты, которые, в свою очередь, затем вводятся псевдобеременным суррогатным матерям. В качестве альтернативы векторы могут быть непосредственно введены в оплодотворенные зиготы, а затем эти зиготы имплантированы суррогатным матерям.Затем проводят наблюдение за потомством на предмет развития лейкемии. D) Модели ксенотрансплантатов. Первичные образцы пациентов или линии клеток человека можно вводить непосредственно животным-хозяевам с ослабленным иммунитетом. Животные-хозяева часто подвергаются воздействию ионизирующего излучения перед инъекцией образца для подавления остаточной иммунной функции.
Вирусные и транспозонные модели
Одной из самых ранних вирусных моделей ОМЛ является эритробластный ОМЛ, вызываемый вирусами лейкемии Френда, о котором впервые сообщила в 1957 году доктор Шарлотта Френд [16].Она сообщила о присутствии бесклеточного агента, который может серийно передавать эритробластный лейкоз у восприимчивых линий мышей. Позднее было обнаружено, что этот фильтруемый агент представляет собой комбинацию вируса, формирующего очаг селезенки с дефицитом репликации (SFFV), и репликационно-компетентного вируса мышиного лейкоза Friend (MuLV) [17]. SFFV был идентифицирован как причина эритробластного лейкоза [18]; он частично действует путем инсерционного мутагенеза (см.). Интеграция вирусного генома в непосредственной близости от Spi / PU.1 ген приводит к его сверхэкспрессии, управляемой вирусным LTR, что способствует снижению дифференцировки эритробластов, наблюдаемой при заболевании [19]. MuLV был использован для обнаружения новых генов, участвующих в лейкемогенезе AML, в скринингах инсерционного мутагенеза [20]. Дополнительные вирусы лейкемии, индуцирующие AML, были выделены и использованы в скринингах инсерционного мутагенеза с использованием трансгенных моделей, включая вирус MOL4070LTR [21–22]. Вирусно-индуцированный ОМЛ также использовался в доклинической оценке возможных терапевтических средств, в первую очередь на модели спонтанной лейкемии у мышей AKR в результате онкогенного РНК-вируса [23].Помимо вирусно-опосредованного инсерционного мутагенеза, также были разработаны системы на основе транспозонов, в первую очередь система Sleeping Beauty, которая использовалась для идентификации взаимодействующих мутаций [24-25]. Эти модели, индуцированные вирусами и транспозонами, способствовали пониманию лейкемогенеза и идентификации активных терапевтических агентов. Однако их отношение к заболеванию человека сомнительно, поскольку не было получено убедительных доказательств наличия у человека вируса или транспозона, индуцирующего ОМЛ.
Трансгенные модели
ОМЛ характеризуется неслучайными повторяющимися кариотипическими аномалиями, которые, как известно, влияют на прогноз, особенно хромосомные транслокации. Эти транслокации могут быть патогномоничными для определенного подтипа AML, такого как t (15; 17), наблюдаемого при остром промиелоцитарном лейкозе (APL), или могут возникать при различных лейкозах, таких как филадельфийская хромосома, наблюдаемая при CML, ALL и редко AML.
Самые ранние трансгенные модели мышей, созданных с помощью AML, для экспрессии слитых белков, генерируемых этими транслокациями.В трансгенных моделях мышей создаются путем манипуляции с эмбриональными стволовыми (ES) клетками. В классическом подходе ДНК вводится непосредственно в про-ядро оплодотворенных зигот, которые затем вводятся псевдобеременным самкам (см.). Это приводит к нецелевой интеграции трансгена и зависит от экспрессии трансгена для генерации фенотипа. В более современном подходе ДНК электропорируется в ES-клетки, а интегранты отбираются путем экспрессии генов устойчивости к антибиотикам.Выбранные клетки затем вводятся в тетраплоидные бластоцисты, которые, в свою очередь, имплантируются псевдобеременным самкам. Затем потомство подвергают обратному скрещиванию с мышами дикого типа для получения гомозиготно трансгенных мышей. Теперь можно создать векторы, которые нацелены на определенные сайты в геноме посредством гомологичной рекомбинации. Дополнительный уровень сложности может быть добавлен с помощью более новых векторов, которые условно экспрессируют гены в ответ на доксициклин (системы включения / выключения Tet) или рекомбиназу Cre (с использованием систем Lox / Cre).Как только мыши с этими условными аллелями созданы, их можно скрещивать с другими трансгенными животными, которые экспрессируют трансактиватор Tet или рекомбиназу Cre тканеспецифическим образом, чтобы генерировать тканеспецифическую экспрессию трансгена (обзор в [26]). В дополнение к экспрессии онкогенных слитых белков, онкогенные аллели с усилением функции могут быть «сбиты» с их соответствующих нормальных локусов, а опухолевые супрессоры могут быть «выбиты» с использованием тех же подходов.
APL
Ранние модели APL были созданы путем экспрессии гибридного белка, генерируемого t (15; 17), PML-RAR, под контролем различных миелоид-специфичных промоторов [27–29].Эти модели не только воспроизводили гистологический фенотип APL человека, но также ремиссии, наблюдаемые после лечения всей транс-ретиноевой кислотой (ATRA). В трансгенной модели, экспрессирующей слитый белок PLZF-RAR, обнаруженной у пациентов с редким вариантом APL, содержащим t (11; 17), лечение ATRA не вызывало ремиссии у мышей, имитируя клинический сценарий [30]. Несмотря на эти успехи, у модели есть несколько недостатков. Результирующий фенотип зависит от экспрессии встроенного трансгена.При использовании различных миелоид-специфичных промоторов некоторые промоторы давали модели с высокой пенетрантностью и более короткими латентными периодами, в то время как другие выявляли заболевание, более напоминающее миелопролиферативные новообразования, чем острый лейкоз. Действительно, модель, экспрессирующая PML-RARα под контролем промотора CD11b, вообще не развивала лейкоз [27]. Эти модели все еще оставляют вопрос о том, необходимы ли генетические события в дополнение к t (15; 17), чтобы вызывать APL у людей.
AML1-ETO Leukemias
Слитый белок AML1-ETO генерируется t (8; 21), обнаруживаемым преимущественно в AML FAB класса M2.Пациенты с ОМЛ, положительные по транслокации t (8; 21), имеют лучший прогноз и, как правило, поддаются лечению химиотерапевтическими препаратами [31–32]. Слитые гены AML-ETO приводят к гибели эмбрионов при вставке в эндогенный промотор AML1 из-за плохого гематопоэза, аналогично AML1 — / — животных с нокаутом [33]. Это делает стандартные модели AML1-ETO неинформативными. Чтобы обойти это ограничение, доктор Чжан и его коллеги создали индуцибельную трансгенную модель, которая экспрессирует AML1-ETO под контролем репрессора Tet.Несмотря на устойчивую экспрессию трансгена в костном мозге мышей, лейкоз не развился [34]. Впоследствии они обнаружили, что когда мышей, экспрессирующих AML1-ETO (под контролем миелоид-специфического промотора MRP8 ), лечили канцерогеном, N-этил-N-нитрозомочевиной, у 55% животных развился AML [35], в то время как ни один из них не развивался. контрольных мышей. Это открытие согласуется с мнением, что экспрессия AML1-ETO необходима, но недостаточна для лейкемогенеза. В соответствии с этой идеей другие группы обнаружили сотрудничество AML1-ETO с мутациями в тирозинкиназах, таких как TEL-PDGFR, используя мозаичные модели, которые будут обсуждаться ниже [36–37].
MLL Leukemias
Ген смешанной лейкемии (MLL) расположен на хромосоме 11 и часто участвует в сбалансированных транслокациях при остром лейкозе. У него более 40 известных партнеров слияния при остром лейкозе, и он часто ассоциируется с химиорезистентностью и плохим прогнозом [31–32, 38]. Примерно 5% пациентов с острым лейкозом экспрессируют слияния MLL [39–40], хотя это число увеличилось до ~ 70% пациентов, ранее подвергавшихся воздействию антрациклинов и ядов топоизомеразы II.Модели слияния MLL-белков, таких как Mll-lacZ, приводят к AML у одних животных, но не у других. LacZ не имеет известной онкогенной роли, поэтому это предполагает, что одного MLL достаточно, чтобы вызвать онкогенез. В сценарии t (9; 11) AF9 сотрудничает с MLL. Белок AF9 гомологичен продукту ENL из 19p13.3, который является другим геном-партнером для MLL, участвующего в острых лейкозах. Corral et al. использовали гомологичную рекомбинацию в ES-клетках для получения слияния Mll-AF9. Слитый белок, который они создали, зависит от эндогенных контрольных элементов MLL для транскрипции слитого белка.У мышей развился ОМЛ, несмотря на широко распространенную активность промотора MLL [41]. У небольшой группы животных развился ОЛЛ, что согласуется с тем, что наблюдается у людей [42]. Инженерия реципрокной транслокации между хромосомами 9 и 11 для генерации слитого гена MLL-AF9 была создана в одной модели с использованием сайтов LoxP как в локусах MLL, так и в AF9, но у этих мышей не развился лейкоз [43]. Модель продемонстрировала возможность использования систем Lox-Cre для создания транслокаций у мышей.Другая рекуррентная транслокация с участием гена MLL происходит с t (11; 19) и приводит к генерации слитого белка MLL-ENL. Это событие слияния было смоделировано на трансгенных животных с использованием подхода инженерной транслокации [44]. В этом исследовании мышей получали с сайтами LoxP как в генах MLL, так и в генах ENL и скрещивали с мышами, экспрессирующими Cre с гена Lmo2 , специфичного для гематопоэтических клеток. Полученные в результате мыши развили агрессивный, полностью пенетрантный AML, демонстрируя, что подход инженерной транслокации может успешно моделировать AML.
Модели усиления функции, «сбитые с толку»
Несколько онкогенов вовлечены в развитие ОМЛ у людей, в первую очередь тирозинкиназа рецептора Flt3. Мутации Flt3 встречаются у 25% пациентов с ОМЛ и имеют худший прогноз [45]. Наиболее распространенная мутация Flt3 включает в себя создание тандемной дупликации в рамке считывания, так называемая мутация «Flt3 ITD». Flt3 ITD является конститутивно активной формой Flt3, которая вызывает лиганд-независимую передачу сигналов. Была сгенерирована встроенная модель ITD Flt3; у полученных мышей развилось миелопролиферативное новообразование, но не AML; согласуется с другими моделями, которые предполагают, что ITD Flt3 может приводить к усилению пролиферации, но одного недостаточно для развития AML [46].Также согласуется с этим открытием, когда трансгенных мышей, экспрессирующих ITD Flt3, скрещивали с трансгенными животными, экспрессирующими слитый белок NUP98-HOXD13, обнаруженный у пациентов с миелодиспластическим синдромом (МДС) и AML, в результате у полученного потомства развился высокопенетрантный и летальный AML [47].
G-белок Ras является мишенью для активации мутаций при AML. K-ras мутирует примерно в 10–15% AML и в 20–25% N-ras. Была разработана индуцибельная модель K-ras, управляемая его эндогенным промотором [48].В этой модели исследователи создали мутированный аллель K-ras ниже транскрипционной стоп-последовательности, фланкированной сайтами LoxP. Затем эту мышь скрестили с мышью, которая экспрессирует рекомбиназу Cre под контролем интерферон-индуцируемого промотора MX-1 (MX1-Cre). Условная экспрессия K-ras G12D приводит к фатальному миелопролиферативному нарушению, аналогичному таковому в моделях ITD Flt3. Этот вывод согласуется с гипотезой «двух ударов», согласно которой необходимы два изменения, чтобы вызвать ОМЛ [2].Интересно, что когда аналогичная модель была построена с использованием N-ras G12D , был индуцирован гораздо более вялый миелопролиферативный ответ, и мыши умерли от ряда гематологических злокачественных новообразований, демонстрируя, что разные аллели Ras не являются функционально эквивалентными [49].
Наиболее частая мутация при ОМЛ находится в гене неуклофосмина 1 или NPM1 . Он выполняет множество клеточных функций и перемещается между ядерными, ядрышковыми и цитоплазматическими локациями. При AML ~ 35% пациентов имеют мутацию NPM1, которая изменяет клеточную локализацию, что приводит к цитоплазматическому распределению только мутантного белка.Первая модель, которая сбивала мутированную форму NPM1 в эндогенный локус мыши, привела к легкому миелопролиферативному нарушению, но не к AML. Во второй модели наиболее распространенная форма мутации NPM1 была вставлена в локусы мыши, фланкированные сайтами LoxP; Полученные мыши были скрещены с мышами MX1-Cre. После индукции Cre примерно у одной трети двойных трансгенных мышей развился ОМЛ с отсроченным началом. Пенетрантность увеличивалась, а латентность сокращалась с использованием подхода инсерционного мутагенеза на основе транспозонов.Эти модели прояснили роль мутаций, которые приводят к онкогенному усилению функции в развитии AML, и, вероятно, будут продолжать вносить вклад в более глубокое понимание молекулярного патогенеза заболевания.
«Нокаутированные» модели с потерей функции
Некоторые пути роста и выживания могут быть гиперактивированы из-за потери отрицательных регуляторов. Одним из таких примеров при AML является путь киназы PI3, который гиперактивируется у 50–70% пациентов с AML [50].Супрессор опухолей PTEN является негативным регулятором этого пути. Когда были получены трансгенные мыши, которые содержали сайты LoxP, фланкирующие PTEN, и этих животных скрестили с штаммом, специфичным для миелоида Cre, у 11 из 18 потомков развился AML. Полученный AML был моноцитарным по морфологии, а путь киназы PI3 был активирован во многих случаях экстрамедуллярного моноцитарного AML [51]. Негативным регулятором пути Ras является ген NF1 , который стимулирует ГТФазную активность белков Ras, приводя к их инактивации.Потеря NF1 была спроектирована путем конструирования локусов NF1, фланкированных сайтами LoxP, когда этих животных скрещивали с индуцибельной моделью K-Ras G12D , описанной выше. При индуцировании Cre образовывался агрессивный AML с высокой пенетрантностью [52]. Эти модели демонстрируют, что трансгенный подход может применяться в сочетании с системами LoxP-Cre для моделирования потери опухолевых супрессоров при генерации AML.
Мозаичные модели
Как обсуждалось выше, хотя мышиные трансгенные модели внесли большой вклад в наше понимание AML, эти модели имеют существенные недостатки.Количество времени и усилий, необходимых для перехода от конструирования переносчиков к скринингу мышей-основателей и поддержанию размножающейся колонии, может быть непомерно высоким. Альтернативный подход к трансгенной модели состоит в том, чтобы взять гемопоэтические стволовые клетки (HSC) от мышей, манипулировать ими ex vivo и трансплантировать их аутологичным или сингенным реципиентам (см.). Эта модель позволяет быстро генерировать генетически определенные лейкозы, чаще всего за счет ретро- или лентивирусной трансдукции клеток (см. Обзор [26]).
Векторы на основе вируса стволовых клеток мыши (MSCV) являются наиболее часто используемыми ретровирусами с дефицитом репликации при создании моделей мозаичного лейкоза. Эта быстрая и экономичная система широко использовалась при ОМЛ в дополнение к исследованиям, проведенным на трансгенных животных. Например, исследователи ретровирусно трансфицировали и трансдуцировали гибридный белок AML1-ETO в HSC для получения химерных мышей с молекулярными аномалиями, схожими с таковыми у пациентов с этой транслокацией [53].Модели трансплантации с использованием костного мозга, трансдуцированного с помощью AML1-ETO и N-ras G12D , в сингенных реципиентов, показали сотрудничество, ведущее к лейкемии, которая напоминала подтип M2 AML, наиболее часто связанный с AML1-ETO [31]. Мозаичная модель AML, созданная слиянием MLL-ENL и N-ras G12D , напоминала подтип M4-M5, связанный со слияниями MLL. Когда лейкозных мышей лечили цитарабином и антрациклиновой химиотерапией, ответы на две модели значительно различались: мыши MLL-ENL были плохо восприимчивы, в то время как мыши AML-ETO показали высокие показатели ремиссии и даже несколько излечений.Эти ответы отражают клиническую ситуацию и демонстрируют, что эти модели могут резюмировать различные прогностические эффекты этих транслокаций. Кроме того, мозаичная модель использовалась для моделирования прогностических эффектов мутировавших тирозинкиназ. При использовании для изучения ответа на стандартные химиотерапевтические препараты AML, Flt3 ITD ускорял приживление. Эта модель была чувствительна к цитарабину, обеспечивая снижение опухолевой нагрузки и улучшение выживаемости. Доксорубицин не показал такой же эффективности, и было высказано предположение о чистой резистентности при сочетании двух препаратов, как при стандартной индукционной терапии ОМЛ [54].
Модели ксенотрансплантатов
Даже самый клинически агрессивный образец лейкозных клеток от пациента вряд ли вырастет при помещении в культуру, а характеристики клеток могут измениться в зависимости от используемой среды [55–56]. Кроме того, системы культивирования не могут воспроизводить взаимодействия между лейкозными клетками и их стромой и иммунными клетками. Чтобы обойти эти ограничения, были разработаны мышиные модели ксенотрансплантатов, позволяющие приживить первичные образцы пациентов и клеточные линии.
В первых попытках ксенотрансплантации ОМЛ мышам с ослабленным иммунитетом использовались бестимусные голые мыши, несущие мутацию в гене Foxn1 , что привело к почти полному отсутствию функциональных Т-клеток. Ранние эксперименты с использованием первичных образцов пациентов действительно привели к образованию гранулоцитарных сарком, но без вовлечения костного мозга и других органов [57]. В попытке улучшить приживление костного мозга мышей с тяжелым комбинированным иммунодефицитом (SCID) тестировали на их способность приживать образцы пациентов с AML.У этих мышей есть мутации в гене Prkdc и, следовательно, отсутствуют функциональные Т- или В-клетки. Хотя образцы, введенные внутрибрюшинно или имплантированные в капсулы почек, имели значительную скорость отбора, внутривенная инъекция привела к приживлению очень небольшого количества образцов у животных-реципиентов [58]. В важной статье доктор Джон Дик и его коллеги вводили первичные образцы пациентов мышам SCID, которых затем лечили фактором стволовых клеток человека (SCF) и фактором, стимулирующим колонии гранулоцитов-макрофагов (GM-CSF).Это привело к высокому количеству заборов периферической крови или костного мозга пациента, введенных внутривенно; что еще более важно, только часть лейкозных клеток могла успешно прижиться [10]. Это было первое свидетельство иерархии лейкозных клеток, при которой меньшинство населения отвечает за поддержание болезни. С тех пор были выведены мыши с более полным иммунодефицитом. У мышей с диабетом без ожирения (NOD) / SCID нет функциональных B- или T-клеток и снижена активность NK-клеток и моноцитов.У них более высокая скорость приживления первичных образцов пациентов, чем у мышей SCID [59]. Дальнейшее ослабление иммунитета было установлено у мышей NOD / SCID, у которых есть делеции в гене, кодирующем гамма-цепь рецептора интерлейкина 2 (IL2Rγ) (так называемые мыши NSG). Наконец, на этой платформе была создана трансгенная мышь, которая экспрессирует человеческий iL3, GM-CSF и SCF (NSG-tg). Эта мышь значительно улучшила скорость приживления первичных образцов пациентов по сравнению со стандартной мышью NSG [60]. Хотя возможности этих моделей ограничены в отношении взаимодействий лейкозных иммунных клеток, они являются ценными инструментами для оценки доклинической активности многообещающих агентов и взаимодействий лейкозных клеток со стромой.
Немышиные модели
Для изучения ОМЛ использовались другие животные модели, включая крыс. В то время как спонтанные лейкемии у крыс редки, сообщалось о многих моделях, вызванных канцерогенами и радиацией [61–64]. Миеломоноцитарный лейкоз, L5222, был индуцирован в 1967 г., через 326 дней после однократной внутривенной инъекции этилнитрозомочевины (200 мг / кг массы тела) трехмесячной самке крысы BD IX. Этот лейкоз был трансплантирован крысам BD IX и использовался в доклинических исследованиях эффективности [65].Модель APL, созданная на крысах Brown Norway (BNML), использовалась в доклинических исследованиях химиотерапии и трансплантации [66]. Лейкоз был индуцирован у самок крыс линии BN 9,10-диметил-1,2-бензантраценом [67] и имеет характеристики образования колоний, аналогичные характеристикам образования человеческих образцов AML в анализах колоний [68]. Кроме того, эта модель внесла важный вклад в понимание минимальной остаточной болезни при ОМЛ (см. Обзор [69]). Совсем недавно появились сообщения о системах AML, не относящихся к млекопитающим.Одним из примеров является недавний отчет о трансплантации образцов AML человека эмбрионам рыбок данио. В этом исследовании исследователи вводили человеческие клеточные линии AML и образцы пациентов в эмбрионы рыбок данио через 48 часов после оплодотворения. Они использовали от 50 до 200 клеток и увидели снижение лейкемической нагрузки, когда эмбрионы, которым вводили чувствительные клетки, обрабатывали иматинибом [70]. В другом исследовании человеческие APL и CML в клеточных линиях миелоидного бластного кризиса вводили эмбрионам рыбок данио; Затем эмбрионы обрабатывали иматинибом или ATRA, и у них отмечалось снижение лейкемической нагрузки [71].
Другой модельной системой, используемой для изучения AML, является плодовая муха, Drosophila melanogaster . Экспрессия слитого белка AML1-ETO в клетках крови Drosophila нарушала дифференцировку клеток крови, которые полагаются на RUNX1, подобно тому, что наблюдается у пациентов [72]. Эти системы, не относящиеся к млекопитающим, обладают преимуществами более низкой стоимости и повышенной репродуктивной способности в системах с хорошо изученной генетикой. Они могут внести значительный вклад в понимание патогенеза ОМЛ.
Модели ВСЕ на животных
ВСЕ — наиболее распространенная лейкемия у детей, а также встречается у значительного числа взрослых. Для него характерно агрессивное разрастание клональных лимфобластов. ОЛЛ имеет несколько подтипов, включая пре-В-клетки, Т-клетки и зрелые В-клетки или клетки Беркитта. Он может проявляться в основном в виде твердой массы (лимфобластные лимфомы) или с первичным поражением костного мозга (лимфобластные лейкемии). В отличие от AML, сейчас считается, что более 90% детей с диагнозом ALL могут быть излечены [73].Хотя это замечательное достижение является результатом многих направлений исследований, несколько важных открытий в отношении ALL было обнаружено с использованием моделей на животных. Эти выводы включают демонстрацию того, что активация Notch-1 приводит к Т-клеточному ОЛЛ на мышиной модели трансплантата [74], открытие, что у трансгенных мышей, экспрессирующих версию слияния BCR-ABL p190, развился ОЛЛ [75], и доклиническую оценку активность антиметаболитов [76].
Мышиные модели
Как и в случае AML, мышиные модели ALL используют канцероген-индуцированный, вирусно-индуцированный, трансгенный, мозаичный и ксенотрансплантный подходы.Эти модели внесли свой вклад в нынешнее понимание ВСЕГО.
Модели, индуцированные канцерогенами
Как и в случае AML, ранние исследования на мышах основывались на модели, индуцированной канцерогеном. В дополнение к важной клеточной линии L1210, обсужденной выше, существует несколько других канцероген-индуцированных линий ALL, включая клеточную линию L5178Y. Эта клетка является производной DBA / 2-производной метилхолантрен-индуцированной Т-клеточной линии, которая широко использовалась в доклинических исследованиях эффективности и механизмов устойчивости [77–79].
Вирусные модели
Штамм мышей AKR, склонный к лейкемии, был использован для характеристики онкогенных свойств различных вирусов. В результате была получена модель Т-клеточного ОЛЛ у мышей AKR, инфицированных рекомбинантным ретровирусом, нацеленным на тимоциты [80]. Эта модель использовалась для оценки влияния ожирения на прогрессирование ОЛЛ [81]. Кроме того, лимфобластные лейкозы и лимфомы, возникшие в результате мутагенеза вставки ретровируса, позволили охарактеризовать новые гены, участвующие в лимфоидном лейкемогенезе, путем характеристики общих последовательностей вставки вируса [82].Надежность методов была подтверждена, когда было обнаружено, что идентифицированный с помощью скрининга ген Prdm14 сверхэкспрессируется в человеческом B- и T-клеточном ОЛЛ и может инициировать ОЛЛ при сверхэкспрессии вирусной трансдукцией клеток костного мозга мыши [83].
Трансгенные модели
Было опубликовано несколько ВСЕ трансгенных моделей. Эти модели воспроизводят В-клетки, Т-клетки и лейкоз Беркитта / лимфобластные лимфомы.
BCR-ABL
Слитый белок BCR-ABL является результатом транслокации t (9; 22) (q34; q11), также известной как филадельфийская хромосома.Эта транслокация характерна для ХМЛ, но также обнаруживается при В-клеточном ОЛЛ. Существует несколько изоформ этого слитого белка, форма p210 (обнаруживается в основном при CML) и форма p190 (обнаруживается в основном при ALL). Были созданы трансгенные мыши, экспрессирующие изоформу p190 с промотора BCR, но у них была эмбриональная летальность [75]. Напротив, трансгенные мыши, экспрессирующие слияние p190 BCR-ABL с промотора металлотионеина, рождаются живыми и 95% умирают от пре-B-клеточного лейкоза / лимфомы через 35–200 дней [84].Эта модель была использована для тестирования активности новых соединений [85], ингибиторов тирозинкиназы [86] и влияния ожирения на прогрессирование ОЛЛ [81].
MLL Leukemias
Как следует из названия, лейкемия смешанного происхождения, транслокации MLL наблюдаются как при ОМЛ, так и при ОЛЛ. Слитый белок MLL-AF4 наблюдается у пациентов с t (4; 11), и он сильно связан с В-клеточным ОЛЛ. Была создана модель трансгенной мыши, которая экспрессировала MLL-AF4 с эндогенного промотора MLL. Полученные мыши продемонстрировали нерегулируемый лимфоидный и миелоидный рост, а после длительного латентного периода — В-клеточные лимфомы [87].Другая модель была создана с использованием инвертированного аллеля AF4, нацеленного на интрон в гене MLL; при воздействии рекомбиназы Cre происходила инверсия и образование MLL-AF4. Затем мышей скрещивали с различными трансгенными мышами с тканеспецифической экспрессией Cre. Когда присутствовал Cre, у мышей развивались злокачественные новообразования В-клеток с различной латентностью в зависимости от промотора Cre [88]. Трансгенная модель с использованием слитого белка MLL-AF4 человека, экспрессированного из вирусного LTR MSCV, привела к В-клеточным новообразованиям, и латентность могла быть сокращена путем скрещивания этих животных с трансгенными мышами, экспрессирующими K-ras G12D [89].Этот результат подтверждает гипотезу множественного попадания в лейкемогенез.
Eµ-myc
Лимфома / лейкемия Беркитта считается подтипом ОЛЛ и характеризуется транслокациями, в результате которых ген MYC экспрессируется под контролем промоторов тяжелой или легкой цепи иммуноглобулина. В результате болезнь характеризуется высокой скоростью распространения и очень агрессивным поведением. Были получены трансгенные мыши с мышиным геном Myc под контролем промотора тяжелой цепи IgG, имитирующего t (8; 14), наблюдаемый в клинических условиях.В результате у мышей развились агрессивные В-клеточные лимфомы и лейкозы, при этом 90% животных умерли в течение первых 5 месяцев жизни [90]. Эта модель была использована для изучения роли взаимодействующих мутаций в лимфоме / лейкемогенезе [91], а также в основополагающей статье о том, как потеря p53 или избыточная экспрессия BCL2 влияет на химиотерапевтический ответ in vivo [92].
NOTCh2
Рецепторы Notch — это трансмембранные рецепторы, которые при активации лигандом расщепляются на внеклеточные и внутриклеточные части.Внутриклеточная часть (ICN) перемещается в ядро, где она взаимодействует с дополнительными белками, что приводит к транскрипционной активации генов-мишеней (обзор в [93]). NOTCh2 был впервые идентифицирован при транслокации, несущей ALL, Т-лимфоциты человека, t (7; 9) (q34; q34.3). Эта транслокация приводит к образованию гена слияния, в котором промотор TCR управляет экспрессией 3 ’конца локуса NOTCh2 , кодируя только ICN и приводя к конститутивной активации NOTCh2 [94].Эта транслокация обнаруживается только примерно в 1% Т-клеточного ОЛЛ. Однако в последующем сообщении от 50 до 60% образцов Т-клеточного ОЛЛ содержали точечные мутации в NOTCh2, , причастные к патогенезу большинства типов Т-клеточного ОЛЛ [95]. Было создано несколько моделей трансгенных мышей для экспрессии активированных аллелей NOTCh2 из Т-клеточных промоторов [96–98]. У этих мышей изменено развитие тимоцитов CD4, CD8 и развиваются злокачественные опухоли Т-клеток. В более поздней модели используется трансгенная мышь, которая условно экспрессирует две общие точечные мутации в NOTCh2 из своего эндогенного промотора с использованием кассеты Lox-Stop-Lox.Эти мыши продемонстрировали ускоренное развитие злокачественных опухолей Т-клеток в системе мутагенеза транспозонов Спящей красавицы [99].
Mosaic Models
Как и в случае AML, ex-vivo манипуляции с HSC были использованы для создания мышиных моделей ALL. MSCV-векторы, сконструированные для экспрессии слитого белка p190 BCR-ABL, широко использовались для создания моделей ALL B-клеток, положительных по филадельфийской хромосоме. Несколько примеров включают исследование эффектов химиотерапии на развитие мутантов BCR-ABL, устойчивых к ингибиторам тирозинкиназы (TKI) [100], роль BCL6 в устойчивости к TKI [101] и роль супрессора опухолей Arf. в развитии В-клеточного ОЛЛ [102].Онкогены слияния MLL также широко использовались для создания мозаичных ВСЕХ моделей. Исследователи объединили мозаичную модель с моделью ксенотрансплантата и, используя человеческие HSC, полученные из пуповинной крови, инфицировали их вектором, экспрессирующим MLL-ENL на основе MSCV, и вводили их сублетально облученным мышам NOD / SCID. У 26 из 29 мышей, которым вводили инъекцию, развился агрессивный В-клеточный ОЛЛ, демонстрирующий, что MLL-ЭНЛ может индуцировать ОЛЛ в клетках человека [103]. В дополнительном исследовании слияние MLL-AF9 могло продуцировать AML или ALL в CD34 + клетках пуповинной крови человека в зависимости от мыши-реципиента (NSG против NSG-tg) [104].Эти исследования установили возможность создания моделей ОЛЛ из человеческих клеток, размножающихся у мышей с ослабленным иммунитетом, и начали разгадывать, каким образом лейкозы с перегруппировкой MLL могут быть миелоидными или лимфоидными.
Модели ксенотрансплантатов
Те же линии мышей с ослабленным иммунитетом, описанные в разделе ксенотрансплантатов в моделях AML, были использованы для проведения доклинических исследований эффективности во ВСЕХ образцах. Кроме того, они распространили модель лейкозных стволовых клеток на ОЛЛ, наблюдая, что не все В- или Т-клеточные ОЛЛ-клетки могут инициировать лейкемию у мышей NOD / SCID или NSG [105–106].
Немышиные модели
Подразделение инбредных крыс Sprague-Dawley имеет высокую частоту Т-клеточных злокачественных новообразований и использовалось в доклинической оценке терапевтических средств [107–108]. В-клеточный ОЛЛ был получен от инбредных морских свинок и был серийно трансплантирован инбредным реципиентам, но не беспородным животным [109]. Существуют также модели ОЛЛ, не относящиеся к млекопитающим, включая трансгенную модель рыбок данио, основанную на экспрессии слитого белка TELAML1, обнаруженного у пациентов с пре-В-клеточным ОЛЛ с t (12; 21) (p13; q22).Исследователи экспрессировали трансген либо повсеместно (с использованием модифицированного промотора актина), либо ограниченным лимфоидом (с использованием промотора Rag2). Только у рыбок данио, которые экспрессировали трансген повсеместно, развивался ОЛЛ после длительного латентного периода [110]. Скрининг химического мутагенеза с использованием трансгенных рыбок данио, которые экспрессируют усиленный зеленый флуоресцентный белок Т-клеточно-специфическим образом, дал несколько линий, которые имеют наследственный Т-клеточный ОЛЛ [111]. Другая модель ОЛЛ Т-клеток была создана у рыбок данио с использованием мышиного гена c-myc , экспрессируемого под контролем промотора Rag2 рыбок данио.У трансгенных рыб развился Т-клеточный ОЛЛ с короткой латентностью [112]. В элегантной модели доктор Лук и его коллеги использовали трансгенных рыбок данио, которые содержали условный аллель Cre, управляемый промотором теплового шока 70, и скрестили его с трансгенной моделью, содержащей мышиный c-myc , управляемый промотором Rag2 , содержащим промежуточная кассета Lox-stop-DS Red-Lox. Полученное потомство можно было поместить в воду при 37 ° C, чтобы вызвать экспрессию Cre. Иссечение стоп-кассеты может сопровождаться флуоресцентной визуализацией для обнаружения потери DS Red и индукции экспрессии EGFP.У этих животных развилась Т-клеточная лимфобластная лимфома (LBL), экспрессирующая EGFP, которая после лечения тепловым шоком прогрессировала до лейкемии [113]. Эта модель позже была использована для исследования генетических событий, которые приводят к превращению LBL в ALL [114]. У рыбок данио хорошо зарекомендовавшая себя генетика, низкая стоимость и высокие показатели воспроизводства, что делает их идеальной системой для проведения генетических скринингов. Вклад этих моделей только начинается.
Животные модели CML
ХМЛ уникален среди лейкозов тем, что существует единственное онкогенное событие, которое вызывает заболевание — слитый белок BCR-ABL, генерируемый филадельфийской хромосомой.Это была первая известная соматическая хромосомная аномалия, напрямую связанная со злокачественным новообразованием, и стало первым доказательством того, что рак является генетическим заболеванием [115]. Заболевание характеризуется аномальной пролиферацией и накоплением зрелых и созревающих миелоидных клеток. Он имеет три различных клинических фазы: хроническую фазу, ускоренную фазу и фазу взрыва. В прошлом ХМЛ можно было вылечить только трансплантацией аллогенных стволовых клеток, но в последние годы лечение произвело революцию с появлением ингибиторов киназы BCR-ABL, таких как иматиниб, нилотиниб и дазатиниб (обзор в [116]).
Животные модели сыграли уникальную роль в исследованиях ХМЛ. Доктор Ван Эттен показал, что изоформа p210 белка BCR-ABL непосредственно вызывает ХМЛ, используя мозаичную модель мыши. Он использовал ретровирусный вектор, который экспрессировал белок p210 и инфицированные мышиные HSC. Когда инфицированные клетки были трансплантированы сингенным реципиентам, у животных развилось миелопролиферативное заболевание, сильно напоминающее ХМЛ человека [117]. С тех пор эти модели широко использовались для оценки многих аспектов ХМЛ, включая трансформирующий потенциал различных изоформ BCR-ABL [118], генетику прогрессирования заболевания [36] и характеристику стволовых клеток ХМЛ [119]. .
CML также моделировали с использованием трансгенных мышей. В одной модели изоформа p210 экспрессировалась из индуцируемого тетрациклином протомера и скрещивалась с трансгенной мышью, которая экспрессировала tet-трансактиватор с промотора CD34, обеспечивая экспрессию p210 в присутствии доксициклина в компартменте HSC. В результате у мышей развилось миелопролиферативное заболевание с тромбоцитозом [120]. Это противоположно B-клеточному ALL, которое они наблюдали, когда p210 экспрессировался в пре-B-клетках [121], демонстрируя, что фенотип лейкозов, индуцированных BCR-ABL, зависит, по крайней мере, частично от клетки-источника.В дополнение к мозаичным и трансгенным моделям были проведены исследования ксенотрансплантатов с использованием образцов пациентов с ХМЛ. В недавнем примере сохранение популяции стволовых клеток ХМЛ у пациентов в цитогенетической ремиссии было продемонстрировано путем приживления клеток мышам NSG [122]. Это исследование доказывает устойчивость этих лейкозных стволовых клеток даже у пациентов, которые получали терапию TKI в течение многих лет. Первичные образцы пациентов и клеточные линии, полученные от пациентов с ХМЛ в различных фазах, были приживлены бестимусным мышам [123], мышам SCID [124] и мышам NOD / SCID [125].Использование мозаичных, трансгенных и ксенотрансплантных моделей дало неоценимую информацию о молекулярной патологии этого лейкоза.
Животные модели ХЛЛ
ХЛЛ является наиболее распространенной из всех лейкозов и, в отличие от ХМЛ, является генетически гетерогенным заболеванием. Это заболевание пожилых людей, характеризующееся клональной пролиферацией иммунодефицитных CD5 + зрелых В-лимфоцитов. Заболевание считается вялотекущим, при этом средняя выживаемость при низкой стадии заболевания составляет более десяти лет, хотя у подгруппы пациентов будет более скоротечное течение [126].Заболевание может иногда трансформироваться в более агрессивную диффузную крупноклеточную В-клеточную лимфому или, реже, в лимфому Ходжкина посредством процесса, называемого трансформацией Рихтера. К настоящему времени описано несколько моделей ХЛЛ на животных. Как и в случае с другими лейкозами, мыши являются наиболее широко используемой моделью.
Одной из самых ранних моделей мышей с ХЛЛ является спонтанно возникшая модель мышей новозеландских черных (NZB). У мышей NZB развивается клональная пролиферация анеуплоидных лимфоцитов CD5 + B. Заболевание может быть пересажено серийно и иногда превращается в крупноклеточную лимфому, соответствующую трансформации Рихтера, наблюдаемой клинически [127].У этих мышей была обнаружена мутация в локусе микроРНК-16; также наблюдаемый в клетках ХЛЛ человека, считается, что он способствует развитию ХЛЛ [128]. В дополнение к этой спонтанной модели было разработано несколько трансгенных моделей. В трансгенной модели TCL1 человеческий ген TCL1 был клонирован ниже промотора E, что привело к ограничению экспрессии B-клеток на высоком уровне. Ген TCL1 имеет повышенную экспрессию при ряде злокачественных новообразований В-клеток, включая ХЛЛ [129].Полученные мыши развивают клональную или олигоклональную пролиферацию лимфоцитов CD5 + B в более позднем возрасте, имитируя болезнь человека [130]. Дополнительное понимание молекулярной патологии ХЛЛ было получено с помощью двойной трансгенной модели TRAF2DN / Bcl-2. Как Bcl-2, так и факторы, ассоциированные с рецептором фактора некроза опухоли (TRAF), сверхэкспрессируются при злокачественных новообразованиях B-клеток, включая CLL [131–132]. В этой модели исследователи скрестили трансгенных животных, экспрессирующих человеческий BCL2 с промотора тяжелой цепи IgG, с животными, экспрессирующими усеченную форму TRAF2.Полученное потомство развивало прогрессивное накопление клональных CD-экспрессирующих В-клеток, что приводило к сокращению выживаемости [133]. Как и в случае с другими лейкозами, сообщалось о распространении образцов ХЛЛ человека у мышей с ослабленным иммунитетом. Полученная от человека линия клеток CLL, устойчивых к флударабину, WSUCLL, может вызывать опухоли при подкожном введении мышам SCID [134]. Эта модель использовалась для доклинических испытаний [135]. Голые мыши также служили реципиентами клеточных линий CLL человека [136]. Наконец, животных NOD / SCID использовали для приживления первичных образцов пациентов [137].Эта модель была использована для изучения эффектов известных прогностических маркеров ХЛЛ на приживление образца; образцы с неблагоприятными факторами прививаются более агрессивно в этой модели [138]. Первые успехи этой модели делают ее очень привлекательной для будущих доклинических исследований эффективности новых агентов.
Резюме и направления на будущее
Животные модели лейкемии были незаменимы в изучении этих разрушительных болезней. Можно привести убедительный аргумент в пользу того, что наше понимание лейкозов больше, чем любое другое злокачественное новообразование, выиграло от этих модельных систем.Большая часть нынешнего понимания и терапевтических подходов были впервые проверены или открыты с использованием этих моделей. В будущем можно легко представить синергию между различными моделями для дальнейшей разработки новых терапевтических целей. Трансгенные и мозаичные модели имеют очевидную полезность для доклинических испытаний эффективности, а также для изучения взаимодействий стромы и иммунных клеток. У моделей ксенотрансплантатов есть уникальное преимущество, заключающееся в том, что они могут исследовать специфическую биологию клеток человека в in vivo, хотя и в среде с ослабленным иммунитетом.Новые системы, не относящиеся к млекопитающим, могут привести к крупномасштабному генетическому скринингу для выявления критических молекулярных путей, которые могут служить следующим поколением терапевтических мишеней, которые будут оценены на мозаичных, трансгенных и ксенотрансплантных моделях и переведены в клинические испытания для улучшения жизни. наших пациентов с лейкемиями.
Благодарности
Авторы выражают благодарность Карен Кляйн за помощь в редактировании рукописи. Поддержка предоставлена Фондом Дуга Коли по исследованиям лейкемии, Фондом Маккея по исследованиям рака, Фондом исследований Лейта и Фондом Фрэнсис П. Тутуилер.
Библиография
1. Сигел Р., Уорд Э, Броули О., Джемаль А. Статистика рака, 2011: Влияние устранения социально-экономических и расовых различий на преждевременную смерть от рака. CA Cancer J Clin. 2011. 61: 212–236. [PubMed] [Google Scholar] 2. Гиллиланд Д.Г., Джордан Коннектикут, Феликс Калифорния. Молекулярные основы лейкемии. Образовательная программа Hematology Am Soc Hematol. 2004: 80–97. [PubMed] [Google Scholar] 3. Licht JD, Sternberg DW. Молекулярная патология острого миелолейкоза. Образовательная программа Hematology Am Soc Hematol.2005: 137–142. [PubMed] [Google Scholar] 4. Ловенберг Б. Острый миелоидный лейкоз: проблема выявления разнообразия болезней. Образовательная программа Hematology Am Soc Hematol. 2008; 2008: 1–11. [PubMed] [Google Scholar] 5. Dohner H, Estey EH, Amadori S и др. Диагностика и лечение острого миелоидного лейкоза у взрослых: рекомендации международной группы экспертов от имени European Leukemia Net. Кровь. 2010. 115: 453–474. [PubMed] [Google Scholar] 6. Нерви Б., Рамирес П., Реттиг М.П. и др. Хемосенсибилизация острого миелоидного лейкоза (ОМЛ) после мобилизации антагонистом CXCR4 AMD3100.Кровь. 2009. 113: 6206–6214. [Бесплатная статья PMC] [PubMed] [Google Scholar] 7. Jaiswal S, Jamieson CH, Pang WW, et al. CD47 активируется циркулирующими гемопоэтическими стволовыми клетками и лейкозными клетками, чтобы избежать фагоцитоза. Клетка. 2009. 138: 271–285. [Бесплатная статья PMC] [PubMed] [Google Scholar] 8. Маджети Р., Чао М.П., Ализаде А.А. и др. CD47 является неблагоприятным прогностическим фактором и мишенью терапевтических антител для стволовых клеток острого миелоидного лейкоза человека. Клетка. 2009. 138: 286–299. [Бесплатная статья PMC] [PubMed] [Google Scholar] 9.Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Лейкозные клетки создают ниши костного мозга, которые нарушают поведение нормальных кроветворных клеток-предшественников. Наука. 2008; 322: 1861–1865. [PubMed] [Google Scholar] 10. Лапидот Т., Сирард С., Формор Дж. И др. Клетка, инициирующая острый миелоидный лейкоз человека после трансплантации мышам SCID. Природа. 1994; 367: 645–648. [PubMed] [Google Scholar] 11. Джордан CT, Гусман ML, Noble M. Раковые стволовые клетки. N Engl J Med. 2006; 355: 1253–1261. [PubMed] [Google Scholar] 12.Skipper HE, Perry S. Кинетика нормальных и лейкозных популяций лейкоцитов и отношение к химиотерапии. Cancer Res. 1970; 30: 1883–1897. [PubMed] [Google Scholar] 13. Закон LW, Таормина V, Бойл PJ. Ответ острых лимфолейкозов на антагонист пуринов 6-меркаптопурин. Ann N Y Acad Sci. 1954. 60: 244–250. [PubMed] [Google Scholar] 14. Маккормак Э., Брюзруд О., Гьертсен БТ. Животные модели острого миелогенного лейкоза — разработка, применение и перспективы на будущее. Лейкемия. 2005; 19: 687–706.[PubMed] [Google Scholar] 15. Шкипер HE, Schabel FM, Jr, Wilcox WS. Экспериментальная оценка потенциальных противораковых агентов. XXI. Планирование приема арабинозилцитозина для использования его специфичности в S-фазе против лейкозных клеток. Cancer Chemother Rep. 1967; 51: 125–165. [PubMed] [Google Scholar] 16. Френд С. Бесклеточная передача у взрослых швейцарских мышей болезни, имеющей характер лейкемии. J Exp Med. 1957; 105: 307–318. [Бесплатная статья PMC] [PubMed] [Google Scholar] 17. Линемейер Д.Л., Менке Дж. Г., Рускетти С. К., Эванс Л. Х., Сколник Е. М..Последовательности гена оболочки, которые кодируют белок gp52 фокусообразующего вируса селезенки, необходимы для индукции пролиферации эритроидных клеток. J Virol. 1982; 43: 223–233. [Бесплатная статья PMC] [PubMed] [Google Scholar] 18. Вольф Л., Рускетти С. Злокачественная трансформация эритроидных клеток in vivo путем введения нереплицирующегося ретровирусного вектора. Наука. 1985; 228: 1549–1552. [PubMed] [Google Scholar] 19. Back J, Dierich A, Bronn C, Kastner P, Chan S. PU.1 определяет способность эритроидных клеток-предшественников к самообновлению.Кровь. 2004. 103: 3615–3623. [PubMed] [Google Scholar] 20. Erkeland SJ, Valkhof M, Heijmans-Antonissen C, et al. Масштабная идентификация генов болезней, вызывающих острый миелоидный лейкоз. J Virol. 2004; 78: 1971–1980. [Бесплатная статья PMC] [PubMed] [Google Scholar] 21. Кауделл Д., Харпер Д.П., Новак Р.Л. и др. Ретровирусный инсерционный мутагенез идентифицирует активацию Zeb2 как новое лейкемогенное событие взаимодействия у трансгенных мышей CALM-AF10. Кровь. 2010; 115: 1194–1203. [Бесплатная статья PMC] [PubMed] [Google Scholar] 22.Slape C, Hartung H, Lin YW, Bies J, Wolff L, Aplan PD. Ретровирусный инсерционный мутагенез идентифицирует гены, которые взаимодействуют с NUP98-HOXD13 во время лейкемической трансформации. Cancer Res. 2007. 67: 5148–5155. [Бесплатная статья PMC] [PubMed] [Google Scholar] 23. Шкипер HE, Schabel FM, младший, трейдер MW, Laster WR., Младший Ответ на терапию спонтанного, первого прохождения и длинных линий прохождения лейкемии AK. Cancer Chemother Rep., 1969; 53: 345–366. [PubMed] [Google Scholar] 24. Василиу Г.С., Купер Дж. Л., Рад Р. и др.Мутантный нуклеофозмин и взаимодействующие пути приводят к возникновению и прогрессированию лейкемии у мышей. Нат Жене. 2011; 43: 470–475. [Бесплатная статья PMC] [PubMed] [Google Scholar] 25. Коллиер Л.С., Адамс Д.Д., Хакетт С.С. и др. Мутагенез спящей красавицы всего тела может вызвать пенетрантную лейкемию / лимфому и редкую глиому высокой степени злокачественности без связанной с этим эмбриональной летальности. Cancer Res. 2009; 69: 8429–8437. [Бесплатная статья PMC] [PubMed] [Google Scholar] 26. Heyer J, Kwong LN, Lowe SW, Chin L. Генно-инженерные модели мышей, не являющиеся зародышевыми линиями, для трансляционных исследований рака.Нат Рев Рак. 2010; 10: 470–480. [Бесплатная статья PMC] [PubMed] [Google Scholar] 27. Early E, Moore MA, Kakizuka A, et al. Трансгенная экспрессия PML / RARalpha нарушает миелопоэз. Proc Natl Acad Sci U S. A. 1996; 93: 7900–7904. [Бесплатная статья PMC] [PubMed] [Google Scholar] 28. Браун Д., Коган С., Лагассе Е. и др. Трансген PMLRARalpha вызывает острый промиелоцитарный лейкоз у мышей. Proc Natl Acad Sci U S. A. 1997; 94: 2551–2556. [Бесплатная статья PMC] [PubMed] [Google Scholar] 29. Гризолано Дж. Л., Весселшмидт Р. Л., Пеличчи П. Г., Лей Т. Дж..Измененное миелоидное развитие и острый лейкоз у трансгенных мышей, экспрессирующих PML-RAR альфа под контролем регуляторных последовательностей катепсина G. Кровь. 1997. 89: 376–387. [PubMed] [Google Scholar] 30. He LZ, Guidez F, Tribioli C и др. Различное взаимодействие PML-RARalpha и PLZF-RARalpha с корепрессорами определяет дифференциальные ответы на RA в APL. Нат Жене. 1998. 18: 126–135. [PubMed] [Google Scholar] 31. Зубер Дж., Радтке И., Парди Т.С. и др. Мышиные модели ОМЛ человека точно предсказывают ответ на химиотерапию.Genes Dev. 2009; 23: 877–889. [Бесплатная статья PMC] [PubMed] [Google Scholar] 32. Гримуэйд Д., Уокер Х., Оливер Ф. и др. Важность диагностической цитогенетики для исходов при ОМЛ: анализ 1612 пациентов, включенных в исследование MRC AML-10. Рабочие группы Совета медицинских исследований по лейкемии взрослых и детей. Кровь. 1998. 92: 2322–2333. [PubMed] [Google Scholar] 33. Окуда Т., Цай З., Ян С. и др. Экспрессия заблокированного гена лейкемии AML1-ETO ингибирует установление нормального дефинитивного гематопоэза и непосредственно генерирует диспластические гематопоэтические предшественники.Кровь. 1998. 91: 3134–3143. [PubMed] [Google Scholar] 34. Роадс К.Л., Хетерингтон С.Дж., Харакава Н. и др. Анализ роли AML1-ETO в лейкемогенезе с использованием модели индуцибельных трансгенных мышей. Кровь. 2000; 96: 2108–2115. [PubMed] [Google Scholar] 35. Юань Ю., Чжоу Л., Миямото Т. и др. Экспрессия AML1-ETO напрямую участвует в развитии острого миелоидного лейкоза при наличии дополнительных мутаций. Proc Natl Acad Sci U S. A. 2001; 98: 10398–10403. [Бесплатная статья PMC] [PubMed] [Google Scholar] 36.Даш А.Б., Уильямс И.Р., Куток Дж. Л. и др. Мышиная модель бластного кризиса ХМЛ, вызванного сотрудничеством между BCR / ABL и NUP98 / HOXA9. Proc Natl Acad Sci U S. A. 2002; 99: 7622–7627. [Бесплатная статья PMC] [PubMed] [Google Scholar] 37. Гризолано Дж. Л., О’Нил Дж., Каин Дж., Томассон М. Х. Активированная рецепторная тирозинкиназа, TEL / PDGFbetaR, взаимодействует с AML1 / ETO, вызывая острый миелоидный лейкоз у мышей. Proc Natl Acad Sci U S. A. 2003; 100: 9506–9511. [Бесплатная статья PMC] [PubMed] [Google Scholar] 38. Lavau C, Du C, Thirman M, Zeleznik-Le N.Связанные с хроматином свойства CBP, слитого с MLL, вызывают миелодиспластический синдром, который перерастает в миелоидный лейкоз. EMBO J. 2000; 19: 4655–4664. [Бесплатная статья PMC] [PubMed] [Google Scholar] 39. Смотреть на. Онкогенные факторы транскрипции при острых лейкозах человека. Наука. 1997; 278: 1059–1064. [PubMed] [Google Scholar] 40. Daser A, Rabbitts TH. Расширение репертуара гена лейкемии смешанного происхождения MLL в лейкемогенезе. Genes Dev. 2004; 18: 965–974. [PubMed] [Google Scholar] 41. Corral J, Lavenir I, Impey H и др.Ген слияния Mll-AF9, полученный путем гомологичной рекомбинации, вызывает острый лейкоз у химерных мышей: метод создания слитых онкогенов. Клетка. 1996. 85: 853–861. [PubMed] [Google Scholar] 42. Стриссель П.Л., Стрик Р., Томек Р.Дж., Роу Б.А., Роули Д.Д., Железник-Ле, штат Нью-Джерси. Структурные свойства ДНК AF9 аналогичны MLL и могут действовать как горячие точки рекомбинации, приводящие к транслокациям MLL / AF9 и лейкемогенезу. Hum Mol Genet. 2000; 9: 1671–1679. [PubMed] [Google Scholar] 43. Коллинз Э.С., Паннелл Р., Симпсон Э.М., Форстер А., Кролик Т.Х.Межхромосомная рекомбинация генов Mll и Af9, опосредованная cre-loxP в развитии мышей. EMBO Rep. 2000; 1: 127–132. [Бесплатная статья PMC] [PubMed] [Google Scholar] 44. Форстер А., Паннелл Р., Дринан Л. Ф. и др. Разработка de novo реципрокных хромосомных транслокаций, связанных с Mll, для репликации первичных событий рака человека. Раковая клетка. 2003. 3: 449–458. [PubMed] [Google Scholar] 45. Стируолт Д.Л., Радич Ю.П. Роль FLT3 в злокачественных заболеваниях кроветворения. Нат Рев Рак. 2003. 3: 650–665. [PubMed] [Google Scholar] 46.Ли Л., Пилото О., Нгуен Х. Б. и др. Включение внутренней тандемной дупликационной мутации в мышиный FLT3 вызывает миелопролиферативное заболевание на мышиной модели. Кровь. 2008; 111: 3849–3858. [Бесплатная статья PMC] [PubMed] [Google Scholar] 47. Гринблатт С., Ли Л., Слейп С и др. Включение мутации FLT3 / ITD кооперируется со слиянием NUP98-HOXD13 для генерации острого миелоидного лейкоза на модели мышей. Кровь. 2012 [Бесплатная статья PMC] [PubMed] [Google Scholar] 48. Чан ИТ, Гиллиланд Д.Г. Онкогенные K-ras в мышиных моделях миелопролиферативного заболевания и острого миелоидного лейкоза.Клеточный цикл. 2004; 3: 536–537. [PubMed] [Google Scholar] 49. Маккензи К.Л., Дольников А., Миллингтон М., Шоунан Ю., Симондс Г. Мутантные N-расы вызывают миелопролиферативные нарушения и апоптоз у мышей с репопуляцией костного мозга. Кровь. 1999; 93: 2043–2056. [PubMed] [Google Scholar] 50. Мартелли AM, Nyakern M, Tabellini G, et al. Сигнальный путь фосфоинозитид-3-киназы / Akt и его терапевтические последствия для острого миелоидного лейкоза человека. Лейкемия. 2006; 20: 911–928. [PubMed] [Google Scholar] 51. Ю Х, Ли Й, Гао С. и др.Соответствующая мышиная модель моноцитарного лейкоза человека посредством Cre / lox-контролируемой миелоид-специфической делеции PTEN. Лейкемия. 2010; 24: 1077–1080. [Бесплатная статья PMC] [PubMed] [Google Scholar] 52. Каттс Б.А., Шегрен А.К., Андерссон К.М. и др. Дефицит Nf1 взаимодействует с онкогенным K-RAS, вызывая у мышей острый миелоидный лейкоз. Кровь. 2009. 114: 3629–3632. [PubMed] [Google Scholar] 53. de Guzman CG, Warren AJ, Zhang Z, et al. Экспансия гемопоэтических стволовых клеток и отчетливые аномалии развития миелоидов на мышиной модели транслокации AML1-ETO.Mol Cell Biol. 2002; 22: 5506–5517. [Бесплатная статья PMC] [PubMed] [Google Scholar] 54. Парди Т.С., Зубер Дж., Лоу SW. Flt3-ITD изменяет химиотерапевтический ответ in vitro и in vivo p53-зависимым образом. Exp Hematol. 2011; 39: 473–485. e474. [Бесплатная статья PMC] [PubMed] [Google Scholar] 55. Bruserud O, Tore Gjertsen B, Brustugun OT и др. Влияние интерлейкина 10 на бластные клетки, полученные от пациентов с острым миелогенным лейкозом. Лейкемия. 1995; 9: 1910–1920. [PubMed] [Google Scholar] 56. Bruserud O, Gjertsen BT, von Volkman HL.Культура in vitro клеток острого миелогенного лейкоза человека (AML) в бессывороточной среде: исследования нативных бластов AML и клеточных линий AML. J Hematother Stem Cell Res. 2000; 9: 923–932. [PubMed] [Google Scholar] 57. Нара Н., Миямото Т. Прямая и серийная трансплантация острого миелоидного лейкоза человека голым мышам. Br J Рак. 1982; 45: 778–782. [Бесплатная статья PMC] [PubMed] [Google Scholar] 58. Sawyers CL, Gishizky ML, Quan S, Golde DW, Witte ON. Распространение бластных миелоидных лейкозов человека у мышей SCID.Кровь. 1992; 79: 2089–2098. [PubMed] [Google Scholar] 59. Ailles LE, Gerhard B, Kawagoe H, Hogge DE. Характеристики роста предшественников острого миелогенного лейкоза, которые инициируют злокачественное кроветворение у мышей, не страдающих ожирением, диабетом / тяжелым комбинированным иммунодефицитом. Кровь. 1999; 94: 1761–1772. [PubMed] [Google Scholar] 60. Вундерлих М., Чоу Ф.С., Линк К.А. и др. Эффективность ксенотрансплантата AML значительно повышается у мышей NOD / SCID-IL2RG, конститутивно экспрессирующих SCF, GM-CSF и IL-3 человека. Лейкемия. 2010; 24: 1785–1788.[Бесплатная статья PMC] [PubMed] [Google Scholar] 61. Svejda J, Kossey P, Hlavayova E, Svec F. Гистологическая картина лейкемии трансплантируемых крыс, вызванная рентгеновским облучением и метилхолантреном. Новообразования. 1958; 5: 123–131. [PubMed] [Google Scholar] 62. Мориучи Т., Оикава Т., Кодама Т., Ямагути Х., Кобаяси Х. Создание и характеристика трансплантируемого миеломоноцитарного лейкоза крыс. Cancer Res. 1983; 43: 5478–5483. [PubMed] [Google Scholar] 63. Иванкович С., Целлер В.Дж. [Лейкемия L 5222 штамма крыс BD IX.Индуцированная этилнитрозомочевиной моноцитарно-миелоидная трансплантируемая форма для цитохимических и химиотерапевтических исследований] Блют. 1974. 28: 288–292. [PubMed] [Google Scholar] 64. Пирсон Дж. У., Чапарас С. Д., Торгерсен Дж. А., Перк К., Чиригос М. А., Шер Н. А.. Эффект лекарственной терапии против гистологически определенного лейкоза крыс. Cancer Res. 1974; 34: 355–361. [PubMed] [Google Scholar] 65. Zeller WJ, Ivankovic S, Schmahl D. Химиотерапия трансплантируемого острого лейкоза L5222 у крыс. Cancer Res. 1975. 35: 1168–1174. [PubMed] [Google Scholar] 66.Hagenbeek A, Martens AC. Эффективность высоких доз циклофосфамида в сочетании с облучением всего тела при лечении острого миелоцитарного лейкоза: исследования на соответствующей модели крыс. Cancer Res. 1983; 43: 408–412. [PubMed] [Google Scholar] 67. Hagenbeek A, Martens AC. Патогенез крысиной модели острого миелоцитарного лейкоза человека. Haematologica. 1980; 65: 293–308. [PubMed] [Google Scholar] 68. ван Беккум Д.В., ван Остером П., Дике К.А. Образование колоний in vitro при трансплантируемых лейкозах крыс по сравнению с острым миелоидным лейкозом человека.Cancer Res. 1976; 36: 941–946. [PubMed] [Google Scholar] 69. Martens AC, van Bekkum DW, Hagenbeek A. Минимальная остаточная болезнь при лейкемии: исследования на животной модели острого миелоцитарного лейкоза (BNML) Int J Cell Cloning. 1990; 8: 27–38. [PubMed] [Google Scholar] 70. Пруво Б., Жакель А., Дроин Н. и др. Ксенотрансплантат лейкозных клеток в эмбрионе рыбок данио для исследования эффективности лекарств. Haematologica. 2011; 96: 612–616. [Бесплатная статья PMC] [PubMed] [Google Scholar] 71. Коркери Д.П., Деллер Г., Берман Дж. Ксенотрансплантация лейкемии в анализе реакции на химиотерапию рыбок данио in vivo.Br J Haematol. 2011; 153: 786–789. [PubMed] [Google Scholar] 72. Osman D, Gobert V, Ponthan F, Heidenreich O, Haenlin M, Waltzer L. Модель дрозофилы идентифицирует кальпаины как модуляторы человеческого лейкемогенного слитого белка AML1-ETO. Proc Natl Acad Sci U S. A. 2009; 106: 12043–12048. [Бесплатная статья PMC] [PubMed] [Google Scholar] 73. Hunger SP, Lu X, Devidas M и др. Улучшение выживаемости детей и подростков с острым лимфобластным лейкозом в период с 1990 по 2005 год: отчет группы детской онкологии.J Clin Oncol. 2012 [Бесплатная статья PMC] [PubMed] [Google Scholar] 74. Груша В.С., Астер Дж. К., Скотт М.Л. и др. Исключительное развитие Т-клеточных новообразований у мышей с трансплантированным костным мозгом, экспрессирующим активированные аллели Notch. J Exp Med. 1996; 183: 2283–2291. [Бесплатная статья PMC] [PubMed] [Google Scholar] 75. Heisterkamp N, Jenster G, ten Hoeve J, Zovich D, Pattengale PK, Groffen J. Острый лейкоз у трансгенных мышей bcr / abl. Природа. 1990; 344: 251–253. [PubMed] [Google Scholar] 76. Law LW, Dunn TB и др. Наблюдения за действием антагониста фолиевой кислоты на трансплантируемые лимфоидные лейкозы у мышей.J Natl Cancer Inst. 1949; 10: 179–192. [PubMed] [Google Scholar] 77. Тренер DL, Уилок Э.Ф. Характеристика фенотипов клеток L5178Y, выделенных во время прогрессирования состояния покоя опухоли у мышей DBA2. Cancer Res. 1984; 44: 2897–2906. [PubMed] [Google Scholar] 78. Нишимура Т., Муто К., Танака Н. Лекарственная чувствительность устойчивой к адриамицину мутантной сублинии клеток лимфобластомы мыши L5178Y. Дж. Антибиотик (Токио) 1978; 31: 493–495. [PubMed] [Google Scholar] 79. Нисимура Т., Сузуки Х., Муто К., Танака Н. Механизм устойчивости к адриамицину в подлинии клеток лимфобластомы мыши L5178Y.Журнал «Антибиотик» (Токио), 1979; 32: 518–522. [PubMed] [Google Scholar] 80. Клойд М.В., Хартли Дж. В., Роу В.П. Лимфомагенность рекомбинантных вирусов мышиного лейкоза, индуцирующих очаговые клетки норки. J Exp Med. 1980; 151: 542–552. [Бесплатная статья PMC] [PubMed] [Google Scholar] 81. Юн Дж. П., Бехан Дж. В., Хейстеркамп Н. и др. Ожирение, вызванное диетой, ускоряет прогрессирование острого лимфобластного лейкоза в двух моделях на мышах. Cancer Prev Res (Phila) 2010; 3: 1259–1264. [Бесплатная статья PMC] [PubMed] [Google Scholar] 82. Weiser KC, Liu B, Hansen GM и др.Ретровирусные вставки в базе данных VISION идентифицируют молекулярные пути лимфолейкоза и лимфомы мышей. Мамм Геном. 2007. 18: 709–722. [Бесплатная статья PMC] [PubMed] [Google Scholar] 83. Деттман Э.Дж., Симко С.Дж., Аянга Б. и др. Prdm14 инициирует лимфобластный лейкоз после увеличения популяции клеток, напоминающих обычные лимфоидные предшественники. Онкоген. 2011; 30: 2859–2873. [Бесплатная статья PMC] [PubMed] [Google Scholar] 84. Groffen J, Voncken JW, Kaartinen V, Morris C, Heisterkamp N. Ph-позитивный лейкоз: модель трансгенных мышей.Лимфома лейка. 1993; 11 (Приложение 1): 19–24. [PubMed] [Google Scholar] 85. Reichert A, Heisterkamp N, Daley GQ, Groffen J. Лечение Bcr / Abl-положительного острого лимфобластного лейкоза у трансгенных мышей P190 с помощью ингибитора фарнезилтрансферазы SCH66336. Кровь. 2001; 97: 1399–1403. [PubMed] [Google Scholar] 86. Каур П., Фельдхан Н., Чжан Б. и др. Лечение нилотинибом на мышиных моделях лимфобластного лейкоза P190 Bcr / Abl. Молочный рак. 2007; 6: 67. [Бесплатная статья PMC] [PubMed] [Google Scholar] 87. Чен В., Ли К., Хадсон В. А., Кумар А., Кирххоф Н., Керси Дж. Х.Модель «нокаута» на мышах Mll-AF4 приводит к нарушению регуляции лимфоидной и миелоидной регуляции и гематологическому злокачественному новообразованию. Кровь. 2006. 108: 669–677. [Бесплатная статья PMC] [PubMed] [Google Scholar] 88. Мецлер М., Форстер А., Паннелл Р. и др. Условная модель опухолевого генеза B-клеток MLL-AF4 с использованием инверторной технологии. Онкоген. 2006; 25: 3093–3103. [PubMed] [Google Scholar] 89. Тамай Х., Мияке К., Такатори М. и др. Активированный белок K-Ras ускоряет индуцированную MLL / AF4 лейкемолимфомогенность человека в модели трансгенных мышей.Лейкемия. 2011; 25: 888–891. [PubMed] [Google Scholar] 90. Харрис А. В., Пинкерт Калифорния, Кроуфорд М., Лэнгдон В. Ю., Бринстер Р. Л., Адамс Дж. М.. Трансгенная мышь E. mu-myc. Модель спонтанной лимфомы с высокой заболеваемостью и лейкемии ранних В-клеток. J Exp Med. 1988. 167: 353–371. [Бесплатная статья PMC] [PubMed] [Google Scholar] 91. Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW. Мутации INK4a / ARF ускоряют лимфомагенез и повышают химиорезистентность, отключая p53. Genes Dev. 1999; 13: 2670–2677.[Бесплатная статья PMC] [PubMed] [Google Scholar] 92. Шмитт К.А., Фридман Дж. С., Янг М. и др. Программа старения, контролируемая p53 и p16INK4a, способствует результату лечения рака. Клетка. 2002. 109: 335–346. [PubMed] [Google Scholar] 93. Grabher C, von Boehmer H, Look AT. Активация Notch 1 в молекулярном патогенезе Т-клеточного острого лимфобластного лейкоза. Нат Рев Рак. 2006; 6: 347–359. [PubMed] [Google Scholar] 94. Эллисен Л.В., Берд Дж., Западный округ Колумбия и др. TAN-1, человеческий гомолог гена notch дрозофилы, разрушается хромосомными транслокациями в Т-лимфобластных новообразованиях.Клетка. 1991; 66: 649–661. [PubMed] [Google Scholar] 95. Вен А. П., Феррандо А. А., Ли В. и др. Активирующие мутации NOTCh2 при остром лимфобластном лейкозе Т-клеток человека. Наука. 2004. 306: 269–271. [PubMed] [Google Scholar] 96. Deftos ML, Huang E, Ojala EW, Forbush KA, Bevan MJ. Передача сигналов Notch2 способствует созреванию тимоцитов CD4 и CD8 SP. Иммунитет. 2000. 13: 73–84. [Бесплатная статья PMC] [PubMed] [Google Scholar] 97. Фаулкс Б.Дж., Роби Е.А. Переоценка влияния активированного Notch2 на развитие CD4 и CD8 Т-клеток.J Immunol. 2002; 169: 1817–1821. [PubMed] [Google Scholar] 98. Priceputu E, Bouallaga I, Zhang Y, et al. Структурно различные лиганд-связывающие или лиганд-независимые мутанты Notch2 являются лейкемогенными, но по-разному влияют на развитие тимоцитов, апоптоз и метастазирование. J Immunol. 2006; 177: 2153–2166. [PubMed] [Google Scholar] 99. Berquam-Vrieze KE, Swing DA, Tessarollo L, Dupuy AJ. Характеристика трансгенных мышей, экспрессирующих ассоциированные с раком варианты человеческого NOTCh2. Бытие. 2012; 50: 112–118. [Бесплатная статья PMC] [PubMed] [Google Scholar] 100.Boulos N, Mulder HL, Calabrese CR, et al. Химиотерапевтические агенты предотвращают появление устойчивых к дазатинибу мутаций киназы BCR-ABL на точной мышиной модели острого лимфобластного лейкоза с положительной хромосомой в Филадельфии. Кровь. 2011. 117: 3585–3595. [Бесплатная статья PMC] [PubMed] [Google Scholar] 101. Duy C, Hurtz C, Shojaee S и др. BCL6 позволяет Ph + клеткам острого лимфобластного лейкоза выжить при ингибировании киназы BCR-ABL1. Природа. 2011; 473: 384–388. [Бесплатная статья PMC] [PubMed] [Google Scholar] 102.Ван П.Й., Янг Ф., Чен С.Ю. и др. Биологические свойства лейкозов, возникающих в результате BCR / ABL-опосредованной трансформации, варьируются в зависимости от онтогенетического происхождения и активности гена p19ARF. Кровь. 2008; 112: 4184–4192. [Бесплатная статья PMC] [PubMed] [Google Scholar] 103. Барабе Ф., Кеннеди Дж. А., Хоуп К. Дж., Дик Дж. Э. Моделирование возникновения и прогрессирования острого лейкоза человека у мышей. Наука. 2007; 316: 600–604. [PubMed] [Google Scholar] 104. Вэй Дж., Вундерлих М., Фокс С. и др. Микроокружение определяет судьбу клонов в человеческой модели лейкемии MLL-AF9.Раковая клетка. 2008. 13: 483–495. [Бесплатная статья PMC] [PubMed] [Google Scholar] 105. le Viseur C, Hotfilder M, Bomken S и др. При остром лимфобластном лейкозе у детей бласты на разных стадиях иммунофенотипического созревания обладают свойствами стволовых клеток. Раковая клетка. 2008; 14: 47–58. [Бесплатная статья PMC] [PubMed] [Google Scholar] 106. Чиу П.П., Цзян Х., Дик Дж. Э. Клетки, инициирующие лейкоз при Т-лимфобластном лейкозе человека, проявляют устойчивость к глюкокортикоидам. Кровь. 2010; 116: 5268–5279. [PubMed] [Google Scholar] 107.Отова Б., Сладка М., Панчак А., Маринов И. Биологические характеристики спонтанных трансплантируемых Т-клеточных лимфом у инбредных крыс Sprague-Dawley / детенышей. Transplant Proc. 1997; 29: 1754–1755. [PubMed] [Google Scholar] 108. Отова Б., Вацлавикова Р., Даниелова В. и др. Эффекты паклитаксела, доцетаксела и их комбинаций на подкожные лимфомы у инбредных крыс Sprague-Dawley / Cub. Eur J Pharm Sci. 2006. 29: 442–450. [PubMed] [Google Scholar] 110. Sabaawy HE, Azuma M, Embree LJ, Tsai HJ, Starost MF, Hickstein DD.TELAML1 трансгенная модель рыбок данио предшественника В-клеточного острого лимфобластного лейкоза. Proc Natl Acad Sci U S. A. 2006; 103: 15166–15171. [Бесплатная статья PMC] [PubMed] [Google Scholar] 111. Фрейзер Дж. К., Микер Н. Д., Руднер Л. и др. Модели наследственных Т-клеточных злокачественных новообразований, установленные при фенотипическом скрининге у рыбок данио. Лейкемия. 2009; 23: 1825–1835. [Бесплатная статья PMC] [PubMed] [Google Scholar] 112. Лангенау Д.М., Травер Д., Феррандо А.А. и др. Myc-индуцированный Т-клеточный лейкоз у трансгенных рыбок данио. Наука. 2003. 299: 887–890.[PubMed] [Google Scholar] 113. Feng H, Langenau DM, Madge JA, et al. Индукция теплового шока Т-клеточной лимфомы / лейкемии у условно регулируемых Cre / lox трансгенных рыбок данио. Br J Haematol. 2007. 138: 169–175. [PubMed] [Google Scholar] 114. Feng H, Stachura DL, White RM и др. Клетки Т-лимфобластной лимфомы экспрессируют высокие уровни BCL2, S1P1 и ICAM1, что приводит к блокаде интравазации опухолевых клеток. Раковая клетка. 2010. 18: 353–366. [Бесплатная статья PMC] [PubMed] [Google Scholar] 115. Nowell PC, Hungerford DA.Хромосомные исследования при лейкемии человека. IV. Миелопролиферативный синдром и другие атипичные миелоидные нарушения. J Natl Cancer Inst. 1962; 29: 911–931. [PubMed] [Google Scholar] 116. Goldman JM. Начальное лечение пациентов с ХМЛ. Образовательная программа Hematology Am Soc Hematol. 2009: 453–460. [PubMed] [Google Scholar] 117. Дейли GQ, Ван Эттен Р.А., Балтимор Д. Индукция хронического миелогенного лейкоза у мышей геном P210bcr / abl филадельфийской хромосомы. Наука. 1990; 247: 824–830. [PubMed] [Google Scholar] 118.Ли С., Илария Р.Л., младший, Миллион РП, Дейли Г.К., Ван Эттен Р.А. Формы P190, P210 и P230 онкогена BCR / ABL вызывают аналогичный хронический миелолейкозоподобный синдром у мышей, но обладают различной лимфоидной лейкемогенной активностью. J Exp Med. 1999; 189: 1399–1412. [Бесплатная статья PMC] [PubMed] [Google Scholar] 119. Zhang H, Li H, Xi HS, Li S. HIF1alpha необходим для поддержания выживания стволовых клеток хронического миелоидного лейкоза. Кровь. 2012; 119: 2595–2607. [Бесплатная статья PMC] [PubMed] [Google Scholar] 120. Huettner CS, Koschmieder S, Iwasaki H, et al.Индуцируемая экспрессия BCR / ABL с использованием регуляторных элементов CD34 человека приводит к мегакариоцитарному миелопролиферативному синдрому. Кровь. 2003. 102: 3363–3370. [PubMed] [Google Scholar] 121. Хюттнер К.С., Чжан П., Ван Эттен Р.А., Тенен Д.Г. Обратимость острого В-клеточного лейкоза, вызванного BCR-ABL1. Нат Жене. 2000; 24: 57–60. [PubMed] [Google Scholar] 122. Чу С., Макдональд Т., Лин А. и др. Сохранение стволовых клеток лейкемии у пациентов с хроническим миелолейкозом в длительной ремиссии при лечении иматинибом.Кровь. 2011. 118: 5565–5572. [Бесплатная статья PMC] [PubMed] [Google Scholar] 123. Lozzio BB, Lozzi CB, Machado E. Линия клеток миелогенного (Ph +) лейкоза человека: трансплантация бестимусным мышам. J Natl Cancer Inst. 1976; 56: 627–629. [PubMed] [Google Scholar] 124. Skorski T, Nieborowska-Skorska M, Nicolaides NC и др. Подавление роста лейкозных клеток Philadelphia1 у мышей антисмысловым олигодезоксинуклеотидом BCR-ABL. Proc Natl Acad Sci U S. A. 1994; 91: 4504–4508. [Бесплатная статья PMC] [PubMed] [Google Scholar] 125.Дацци Ф., Хассерджиан Р., Гордон М.Ю. и др. Гемопоэтические клетки ХМЛ нормальной и хронической фаз повторно заселяют костный мозг NOD / SCID с различной кинетикой и представлением клеточных клонов. Hematol J. 2000; 1: 307–315. [PubMed] [Google Scholar] 126. Dohner H, Stilgenbauer S, Benner A и др. Геномные аберрации и выживаемость при хроническом лимфолейкозе. N Engl J Med. 2000; 343: 1910–1916. [PubMed] [Google Scholar] 127. Филлипс Дж. А., Мехта К., Фернандес С., Равече Е. С.. Мышь NZB как модель хронического лимфолейкоза.Cancer Res. 1992; 52: 437–443. [PubMed] [Google Scholar] 128. Равече Э.С., Салерно Э., Скаглионе Б.Дж. и др. Аномальный локус микроРНК-16 с синтенией человеческого 13q14, связанный с ХЛЛ у мышей NZB. Кровь. 2007; 109: 5079–5086. [Бесплатная статья PMC] [PubMed] [Google Scholar] 129. Нардуччи М.Г., Пескармона Э., Лазцери С. и др. Регуляция экспрессии TCL1 в B- и T-клеточных лимфомах и реактивных лимфоидных тканях. Cancer Res. 2000; 60: 2095–2100. [PubMed] [Google Scholar] 130. Бичи Р., Шинтон С.А., Мартин Э.С. и др. Хронический лимфоцитарный лейкоз человека, моделируемый на мышах с помощью направленной экспрессии TCL1.Proc Natl Acad Sci U S. A. 2002; 99: 6955–6960. [Бесплатная статья PMC] [PubMed] [Google Scholar] 131. Агилар-Сантелизес М., Роттенберг М.Э., Левин Н., Меллштедт Н., Йондал М. Экспрессия Bcl-2, Bax и p53 в B-CLL в зависимости от выживаемости и клинического прогрессирования in vitro. Int J Cancer. 1996; 69: 114–119. [PubMed] [Google Scholar] 132. Munzert G, Kirchner D, Stobbe H, et al. Сверхэкспрессия гена фактора некроза опухоли, связанного с рецептором фактора 1, при В-клеточном хроническом лимфоцитарном лейкозе: анализ ингибиторов апоптоза, регулируемых NF-каппа B / Rel.Кровь. 2002; 100: 3749–3756. [PubMed] [Google Scholar] 133. Сапата Дж. М., Краевская М., Морс ХК, 3-й, Цой Й., Рид Дж. Домен TNF-рецепторассоциированного фактора (TRAF) и Bcl-2 взаимодействуют, вызывая малую В-клеточную лимфому / хронический лимфоцитарный лейкоз у трансгенных мышей. Proc Natl Acad Sci U S. A. 2004; 101: 16600–16605. [Бесплатная статья PMC] [PubMed] [Google Scholar] 134. Мохаммад Р.М., Мохамед А.Н., Хамдан М.Ю. и др. Создание модели ксенотрансплантата B-CLL человека: полезность в качестве доклинической терапевтической модели. Лейкемия.1996. 10: 130–137. [PubMed] [Google Scholar] 135. Мохаммад Р.М., Лимварапус С., Хамди Н. и др. Обработка модели ксенотрансплантата de novo устойчивой к флударабину ХЛЛ бриостатином 1 с последующим введением флударабина. Int J Oncol. 1999; 14: 945–950. [PubMed] [Google Scholar] 136. Loisel S, Ster KL, Quintin-Roue I, et al. Создание новой модели человеческого B-CLL-подобного ксенотрансплантата на мышах nude. Leuk Res. 2005; 29: 1347–1352. [PubMed] [Google Scholar] 137. Дуриг Дж., Эбелинг П., Грабеллус Ф. и др. Новая модель ксенотрансплантата, не связанного с ожирением, диабетом и тяжелым иммунодефицитом для хронического лимфолейкоза, отражает важные клинические характеристики заболевания.Cancer Res. 2007; 67: 8653–8661. [PubMed] [Google Scholar] 138. Aydin S, Grabellus F, Eisele L, et al. Изучение роли CD38 и функционально связанных молекулярных факторов риска в модели ксенотрансплантата CLL NOD / SCID. Eur J Haematol. 2011; 87: 10–19. [PubMed] [Google Scholar]Что-нибудь ближе к реальности?
Метастаз рака Ред. Авторская рукопись; доступно в PMC 2014 1 июня.
Опубликован в окончательной отредактированной форме как:
PMCID: PMC3568447
NIHMSID: NIHMS416371
Раздел по гематологии и онкологии, Wake Forest Baptist Health, Уинстон-Сейлем, другие статьи в ЧВК, цитирующих опубликованную статью.
Abstract
Модели на животных сыграли неоценимую роль в усилиях по лучшему пониманию и лечению пациентов, страдающих лейкемией. Несмотря на то, что на основе этих моделей были сделаны важные выводы, следует признать ограничения. В этом обзоре мы выделим различные модели лейкемии на животных и опишем их вклад в улучшение понимания и лечения этих видов рака.
Введение
Лейкемии представляют собой разнообразную группу заболеваний, которые приводят к значительной заболеваемости и смертности.В 2011 году у более 44 600 американцев была диагностирована лейкемия, что привело к 21 780 смертельным исходам [1]. Четыре основных типа составляют 85% всех лейкозов: острый миелоидный лейкоз (AML), острый лимфобластный лейкоз (ALL), хронический миелоидный лейкоз (CML) и хронический лимфолейкоз (CLL). Этим и будет посвящен этот обзор. ХЛЛ является наиболее распространенным заболеванием, но на ОМЛ приходится ~ 42% всех смертей от лейкемии [1]. В результате ОМЛ является наиболее интенсивно изучаемым лейкозом и имеет самую разнообразную группу моделей на животных.Трудно переоценить влияние животных моделей на исследования лейкемии. Начиная с новаторской работы доктора Ллойда Лоу о реакции мышиного лимфоидного лейкоза на антиметаболиты в 1940-х и 1950-х годах до открытия лейкемической стволовой клетки доктором Джоном Диком в 1990-х годах, животные модели были неотъемлемой частью нашего понимания биология лейкозов.
Модели ОМЛ на животных
Введение
За последние 40 лет наше понимание молекулярной патофизиологии ОМЛ значительно выросло (обзор в [2–4]).Заболевание проявляется увеличением незрелых, клональных, миелоидных предшественников, что приводит к прогрессирующей недостаточности костного мозга и, в конечном итоге, к смерти. Несмотря на лучшее понимание генетики ОМЛ, терапия для большинства пациентов осталась неизменной, а общая 5-летняя выживаемость составляет 30-40% [5]. Этот неутешительный показатель выживаемости вдохновил на большую работу по разработке более подходящих моделей на животных для использования при обнаружении новых целей и тестировании новых методов лечения. В результате несколько исследований выявили новые идеи, которые стали возможными только благодаря использованию моделей на животных.Несколько примечательных примеров включают роль взаимодействия клеток AML / стромы костного мозга в устойчивости к химиотерапии [6], способность иммунной системы взаимодействовать с AML и нацеливаться на них [7-8], манипулирование нишей гемопоэтических стволовых клеток посредством AML [9], а также плодотворное наблюдение, что AML существует как иерархия клеток с редкой популяцией лейкемических стволовых клеток [10]. Это наблюдение стало центром теории раковых стволовых клеток, которая теперь распространяется и на многие солидные опухоли (см. Обзор [11]).
Мышиные модели
С 1930-х гг. Мышиные модели наиболее широко использовались для изучения ОМЛ. Первоначальные трансплантируемые модели, индуцированные канцерогенами, уступили место более современным трансгенным, ксенотрансплантатным и мозаичным подходам. В этом разделе будут рассмотрены основные типы мышиных моделей AML.
Канцероген-индуцированные модели
Среди первых описанных моделей мышиного лейкоза были трансплантируемые канцерогенные модели, используемые для тестирования возможных терапевтических агентов (см.).Использование антиметаболитных агентов было впервые протестировано на этих моделях. У них были преимущества, заключающиеся в том, что они могли размножать in vitro и затем вводить их большим когортам реципиентов, у которых впоследствии разовьется лейкемия, что позволило провести испытания многообещающих агентов и графиков. Они были использованы с большим эффектом докторами Скиппером и Шабелем для описания кинетики лейкемогенеза и ответа на терапию, которые остаются основной опорой нашего современного понимания болезни (см. Обзор в [12]).Одной из наиболее широко используемых моделей является линия клеток L1210, выделенная доктором Лоу путем воздействия на мышей DBA / 2 канцерогенного 3-метилхолантрена (3-MC) [13]. Полученный лейкоз был получен от умирающего животного и десятилетиями использовался экспериментально. Дополнительные химически индуцированные линии клеток мышиного лейкоза включают, среди прочего, P388, P1534 и L5178Y (обзор см. В [14]). Однако у этих моделей есть несколько недостатков. Самым сильным из них является тот факт, что патогенез этого лейкоза не имеет отношения к большинству случаев ОМЛ у людей, поскольку у немногих людей лейкоз развивается после воздействия химических агентов.Действительно, по большей части считалось, что некоторые из этих клеточных линий более тесно связаны с лимфоидными злокачественными новообразованиями, чем ОМЛ. Кроме того, генетические основы болезни неизвестны, что ограничивает применимость результатов с использованием этих моделей. Несмотря на эти недостатки, эти модели внесли значительный вклад в лечение ОМЛ, особенно в поддержку использования цитарабина [15], который остается одним из наиболее широко используемых агентов для лечения ОМЛ и ОЛЛ.
Основные методы создания моделей лейкемии на животных.А) Модели, индуцированные канцерогеном. Животные из восприимчивых штаммов подвергаются воздействию канцерогенных химических веществ или ионизирующего излучения. После заражения животные наблюдаются на предмет развития лейкемии. Б) Мозаика, модели транспозонов и вирусов. В мозаичных моделях HSC собирают от мышей-доноров и заражают вирусами, кодирующими интересующий онкоген. Интеграция вируса в геном хозяина приводит к доставке и экспрессии онкогена. В дополнение к доставке онкогенов внедрение вируса может приводить к аберрантной экспрессии генов клеточных протоонкогенов (как показано) или к нарушению клеточного супрессора опухоли, если интеграция происходит внутригенно.Мутагенез транспозонов является результатом аналогичных последствий интеграции транспозонов в геном хозяина. В) Трансгенные модели. Направляющий вектор, который был сконструирован для экспрессии интересующего трансгена, инъецировали или электропорировали в мышиные ES-клетки. ES-клетки, которые интегрировали вектор, затем вводятся в тетраплоидные бластоцисты, которые, в свою очередь, затем вводятся псевдобеременным суррогатным матерям. В качестве альтернативы векторы могут быть непосредственно введены в оплодотворенные зиготы, а затем эти зиготы имплантированы суррогатным матерям.Затем проводят наблюдение за потомством на предмет развития лейкемии. D) Модели ксенотрансплантатов. Первичные образцы пациентов или линии клеток человека можно вводить непосредственно животным-хозяевам с ослабленным иммунитетом. Животные-хозяева часто подвергаются воздействию ионизирующего излучения перед инъекцией образца для подавления остаточной иммунной функции.
Вирусные и транспозонные модели
Одной из самых ранних вирусных моделей ОМЛ является эритробластный ОМЛ, вызываемый вирусами лейкемии Френда, о котором впервые сообщила в 1957 году доктор Шарлотта Френд [16].Она сообщила о присутствии бесклеточного агента, который может серийно передавать эритробластный лейкоз у восприимчивых линий мышей. Позднее было обнаружено, что этот фильтруемый агент представляет собой комбинацию вируса, формирующего очаг селезенки с дефицитом репликации (SFFV), и репликационно-компетентного вируса мышиного лейкоза Friend (MuLV) [17]. SFFV был идентифицирован как причина эритробластного лейкоза [18]; он частично действует путем инсерционного мутагенеза (см.). Интеграция вирусного генома в непосредственной близости от Spi / PU.1 ген приводит к его сверхэкспрессии, управляемой вирусным LTR, что способствует снижению дифференцировки эритробластов, наблюдаемой при заболевании [19]. MuLV был использован для обнаружения новых генов, участвующих в лейкемогенезе AML, в скринингах инсерционного мутагенеза [20]. Дополнительные вирусы лейкемии, индуцирующие AML, были выделены и использованы в скринингах инсерционного мутагенеза с использованием трансгенных моделей, включая вирус MOL4070LTR [21–22]. Вирусно-индуцированный ОМЛ также использовался в доклинической оценке возможных терапевтических средств, в первую очередь на модели спонтанной лейкемии у мышей AKR в результате онкогенного РНК-вируса [23].Помимо вирусно-опосредованного инсерционного мутагенеза, также были разработаны системы на основе транспозонов, в первую очередь система Sleeping Beauty, которая использовалась для идентификации взаимодействующих мутаций [24-25]. Эти модели, индуцированные вирусами и транспозонами, способствовали пониманию лейкемогенеза и идентификации активных терапевтических агентов. Однако их отношение к заболеванию человека сомнительно, поскольку не было получено убедительных доказательств наличия у человека вируса или транспозона, индуцирующего ОМЛ.
Трансгенные модели
ОМЛ характеризуется неслучайными повторяющимися кариотипическими аномалиями, которые, как известно, влияют на прогноз, особенно хромосомные транслокации. Эти транслокации могут быть патогномоничными для определенного подтипа AML, такого как t (15; 17), наблюдаемого при остром промиелоцитарном лейкозе (APL), или могут возникать при различных лейкозах, таких как филадельфийская хромосома, наблюдаемая при CML, ALL и редко AML.
Самые ранние трансгенные модели мышей, созданных с помощью AML, для экспрессии слитых белков, генерируемых этими транслокациями.В трансгенных моделях мышей создаются путем манипуляции с эмбриональными стволовыми (ES) клетками. В классическом подходе ДНК вводится непосредственно в про-ядро оплодотворенных зигот, которые затем вводятся псевдобеременным самкам (см.). Это приводит к нецелевой интеграции трансгена и зависит от экспрессии трансгена для генерации фенотипа. В более современном подходе ДНК электропорируется в ES-клетки, а интегранты отбираются путем экспрессии генов устойчивости к антибиотикам.Выбранные клетки затем вводятся в тетраплоидные бластоцисты, которые, в свою очередь, имплантируются псевдобеременным самкам. Затем потомство подвергают обратному скрещиванию с мышами дикого типа для получения гомозиготно трансгенных мышей. Теперь можно создать векторы, которые нацелены на определенные сайты в геноме посредством гомологичной рекомбинации. Дополнительный уровень сложности может быть добавлен с помощью более новых векторов, которые условно экспрессируют гены в ответ на доксициклин (системы включения / выключения Tet) или рекомбиназу Cre (с использованием систем Lox / Cre).Как только мыши с этими условными аллелями созданы, их можно скрещивать с другими трансгенными животными, которые экспрессируют трансактиватор Tet или рекомбиназу Cre тканеспецифическим образом, чтобы генерировать тканеспецифическую экспрессию трансгена (обзор в [26]). В дополнение к экспрессии онкогенных слитых белков, онкогенные аллели с усилением функции могут быть «сбиты» с их соответствующих нормальных локусов, а опухолевые супрессоры могут быть «выбиты» с использованием тех же подходов.
APL
Ранние модели APL были созданы путем экспрессии гибридного белка, генерируемого t (15; 17), PML-RAR, под контролем различных миелоид-специфичных промоторов [27–29].Эти модели не только воспроизводили гистологический фенотип APL человека, но также ремиссии, наблюдаемые после лечения всей транс-ретиноевой кислотой (ATRA). В трансгенной модели, экспрессирующей слитый белок PLZF-RAR, обнаруженной у пациентов с редким вариантом APL, содержащим t (11; 17), лечение ATRA не вызывало ремиссии у мышей, имитируя клинический сценарий [30]. Несмотря на эти успехи, у модели есть несколько недостатков. Результирующий фенотип зависит от экспрессии встроенного трансгена.При использовании различных миелоид-специфичных промоторов некоторые промоторы давали модели с высокой пенетрантностью и более короткими латентными периодами, в то время как другие выявляли заболевание, более напоминающее миелопролиферативные новообразования, чем острый лейкоз. Действительно, модель, экспрессирующая PML-RARα под контролем промотора CD11b, вообще не развивала лейкоз [27]. Эти модели все еще оставляют вопрос о том, необходимы ли генетические события в дополнение к t (15; 17), чтобы вызывать APL у людей.
AML1-ETO Leukemias
Слитый белок AML1-ETO генерируется t (8; 21), обнаруживаемым преимущественно в AML FAB класса M2.Пациенты с ОМЛ, положительные по транслокации t (8; 21), имеют лучший прогноз и, как правило, поддаются лечению химиотерапевтическими препаратами [31–32]. Слитые гены AML-ETO приводят к гибели эмбрионов при вставке в эндогенный промотор AML1 из-за плохого гематопоэза, аналогично AML1 — / — животных с нокаутом [33]. Это делает стандартные модели AML1-ETO неинформативными. Чтобы обойти это ограничение, доктор Чжан и его коллеги создали индуцибельную трансгенную модель, которая экспрессирует AML1-ETO под контролем репрессора Tet.Несмотря на устойчивую экспрессию трансгена в костном мозге мышей, лейкоз не развился [34]. Впоследствии они обнаружили, что когда мышей, экспрессирующих AML1-ETO (под контролем миелоид-специфического промотора MRP8 ), лечили канцерогеном, N-этил-N-нитрозомочевиной, у 55% животных развился AML [35], в то время как ни один из них не развивался. контрольных мышей. Это открытие согласуется с мнением, что экспрессия AML1-ETO необходима, но недостаточна для лейкемогенеза. В соответствии с этой идеей другие группы обнаружили сотрудничество AML1-ETO с мутациями в тирозинкиназах, таких как TEL-PDGFR, используя мозаичные модели, которые будут обсуждаться ниже [36–37].
MLL Leukemias
Ген смешанной лейкемии (MLL) расположен на хромосоме 11 и часто участвует в сбалансированных транслокациях при остром лейкозе. У него более 40 известных партнеров слияния при остром лейкозе, и он часто ассоциируется с химиорезистентностью и плохим прогнозом [31–32, 38]. Примерно 5% пациентов с острым лейкозом экспрессируют слияния MLL [39–40], хотя это число увеличилось до ~ 70% пациентов, ранее подвергавшихся воздействию антрациклинов и ядов топоизомеразы II.Модели слияния MLL-белков, таких как Mll-lacZ, приводят к AML у одних животных, но не у других. LacZ не имеет известной онкогенной роли, поэтому это предполагает, что одного MLL достаточно, чтобы вызвать онкогенез. В сценарии t (9; 11) AF9 сотрудничает с MLL. Белок AF9 гомологичен продукту ENL из 19p13.3, который является другим геном-партнером для MLL, участвующего в острых лейкозах. Corral et al. использовали гомологичную рекомбинацию в ES-клетках для получения слияния Mll-AF9. Слитый белок, который они создали, зависит от эндогенных контрольных элементов MLL для транскрипции слитого белка.У мышей развился ОМЛ, несмотря на широко распространенную активность промотора MLL [41]. У небольшой группы животных развился ОЛЛ, что согласуется с тем, что наблюдается у людей [42]. Инженерия реципрокной транслокации между хромосомами 9 и 11 для генерации слитого гена MLL-AF9 была создана в одной модели с использованием сайтов LoxP как в локусах MLL, так и в AF9, но у этих мышей не развился лейкоз [43]. Модель продемонстрировала возможность использования систем Lox-Cre для создания транслокаций у мышей.Другая рекуррентная транслокация с участием гена MLL происходит с t (11; 19) и приводит к генерации слитого белка MLL-ENL. Это событие слияния было смоделировано на трансгенных животных с использованием подхода инженерной транслокации [44]. В этом исследовании мышей получали с сайтами LoxP как в генах MLL, так и в генах ENL и скрещивали с мышами, экспрессирующими Cre с гена Lmo2 , специфичного для гематопоэтических клеток. Полученные в результате мыши развили агрессивный, полностью пенетрантный AML, демонстрируя, что подход инженерной транслокации может успешно моделировать AML.
Модели усиления функции, «сбитые с толку»
Несколько онкогенов вовлечены в развитие ОМЛ у людей, в первую очередь тирозинкиназа рецептора Flt3. Мутации Flt3 встречаются у 25% пациентов с ОМЛ и имеют худший прогноз [45]. Наиболее распространенная мутация Flt3 включает в себя создание тандемной дупликации в рамке считывания, так называемая мутация «Flt3 ITD». Flt3 ITD является конститутивно активной формой Flt3, которая вызывает лиганд-независимую передачу сигналов. Была сгенерирована встроенная модель ITD Flt3; у полученных мышей развилось миелопролиферативное новообразование, но не AML; согласуется с другими моделями, которые предполагают, что ITD Flt3 может приводить к усилению пролиферации, но одного недостаточно для развития AML [46].Также согласуется с этим открытием, когда трансгенных мышей, экспрессирующих ITD Flt3, скрещивали с трансгенными животными, экспрессирующими слитый белок NUP98-HOXD13, обнаруженный у пациентов с миелодиспластическим синдромом (МДС) и AML, в результате у полученного потомства развился высокопенетрантный и летальный AML [47].
G-белок Ras является мишенью для активации мутаций при AML. K-ras мутирует примерно в 10–15% AML и в 20–25% N-ras. Была разработана индуцибельная модель K-ras, управляемая его эндогенным промотором [48].В этой модели исследователи создали мутированный аллель K-ras ниже транскрипционной стоп-последовательности, фланкированной сайтами LoxP. Затем эту мышь скрестили с мышью, которая экспрессирует рекомбиназу Cre под контролем интерферон-индуцируемого промотора MX-1 (MX1-Cre). Условная экспрессия K-ras G12D приводит к фатальному миелопролиферативному нарушению, аналогичному таковому в моделях ITD Flt3. Этот вывод согласуется с гипотезой «двух ударов», согласно которой необходимы два изменения, чтобы вызвать ОМЛ [2].Интересно, что когда аналогичная модель была построена с использованием N-ras G12D , был индуцирован гораздо более вялый миелопролиферативный ответ, и мыши умерли от ряда гематологических злокачественных новообразований, демонстрируя, что разные аллели Ras не являются функционально эквивалентными [49].
Наиболее частая мутация при ОМЛ находится в гене неуклофосмина 1 или NPM1 . Он выполняет множество клеточных функций и перемещается между ядерными, ядрышковыми и цитоплазматическими локациями. При AML ~ 35% пациентов имеют мутацию NPM1, которая изменяет клеточную локализацию, что приводит к цитоплазматическому распределению только мутантного белка.Первая модель, которая сбивала мутированную форму NPM1 в эндогенный локус мыши, привела к легкому миелопролиферативному нарушению, но не к AML. Во второй модели наиболее распространенная форма мутации NPM1 была вставлена в локусы мыши, фланкированные сайтами LoxP; Полученные мыши были скрещены с мышами MX1-Cre. После индукции Cre примерно у одной трети двойных трансгенных мышей развился ОМЛ с отсроченным началом. Пенетрантность увеличивалась, а латентность сокращалась с использованием подхода инсерционного мутагенеза на основе транспозонов.Эти модели прояснили роль мутаций, которые приводят к онкогенному усилению функции в развитии AML, и, вероятно, будут продолжать вносить вклад в более глубокое понимание молекулярного патогенеза заболевания.
«Нокаутированные» модели с потерей функции
Некоторые пути роста и выживания могут быть гиперактивированы из-за потери отрицательных регуляторов. Одним из таких примеров при AML является путь киназы PI3, который гиперактивируется у 50–70% пациентов с AML [50].Супрессор опухолей PTEN является негативным регулятором этого пути. Когда были получены трансгенные мыши, которые содержали сайты LoxP, фланкирующие PTEN, и этих животных скрестили с штаммом, специфичным для миелоида Cre, у 11 из 18 потомков развился AML. Полученный AML был моноцитарным по морфологии, а путь киназы PI3 был активирован во многих случаях экстрамедуллярного моноцитарного AML [51]. Негативным регулятором пути Ras является ген NF1 , который стимулирует ГТФазную активность белков Ras, приводя к их инактивации.Потеря NF1 была спроектирована путем конструирования локусов NF1, фланкированных сайтами LoxP, когда этих животных скрещивали с индуцибельной моделью K-Ras G12D , описанной выше. При индуцировании Cre образовывался агрессивный AML с высокой пенетрантностью [52]. Эти модели демонстрируют, что трансгенный подход может применяться в сочетании с системами LoxP-Cre для моделирования потери опухолевых супрессоров при генерации AML.
Мозаичные модели
Как обсуждалось выше, хотя мышиные трансгенные модели внесли большой вклад в наше понимание AML, эти модели имеют существенные недостатки.Количество времени и усилий, необходимых для перехода от конструирования переносчиков к скринингу мышей-основателей и поддержанию размножающейся колонии, может быть непомерно высоким. Альтернативный подход к трансгенной модели состоит в том, чтобы взять гемопоэтические стволовые клетки (HSC) от мышей, манипулировать ими ex vivo и трансплантировать их аутологичным или сингенным реципиентам (см.). Эта модель позволяет быстро генерировать генетически определенные лейкозы, чаще всего за счет ретро- или лентивирусной трансдукции клеток (см. Обзор [26]).
Векторы на основе вируса стволовых клеток мыши (MSCV) являются наиболее часто используемыми ретровирусами с дефицитом репликации при создании моделей мозаичного лейкоза. Эта быстрая и экономичная система широко использовалась при ОМЛ в дополнение к исследованиям, проведенным на трансгенных животных. Например, исследователи ретровирусно трансфицировали и трансдуцировали гибридный белок AML1-ETO в HSC для получения химерных мышей с молекулярными аномалиями, схожими с таковыми у пациентов с этой транслокацией [53].Модели трансплантации с использованием костного мозга, трансдуцированного с помощью AML1-ETO и N-ras G12D , в сингенных реципиентов, показали сотрудничество, ведущее к лейкемии, которая напоминала подтип M2 AML, наиболее часто связанный с AML1-ETO [31]. Мозаичная модель AML, созданная слиянием MLL-ENL и N-ras G12D , напоминала подтип M4-M5, связанный со слияниями MLL. Когда лейкозных мышей лечили цитарабином и антрациклиновой химиотерапией, ответы на две модели значительно различались: мыши MLL-ENL были плохо восприимчивы, в то время как мыши AML-ETO показали высокие показатели ремиссии и даже несколько излечений.Эти ответы отражают клиническую ситуацию и демонстрируют, что эти модели могут резюмировать различные прогностические эффекты этих транслокаций. Кроме того, мозаичная модель использовалась для моделирования прогностических эффектов мутировавших тирозинкиназ. При использовании для изучения ответа на стандартные химиотерапевтические препараты AML, Flt3 ITD ускорял приживление. Эта модель была чувствительна к цитарабину, обеспечивая снижение опухолевой нагрузки и улучшение выживаемости. Доксорубицин не показал такой же эффективности, и было высказано предположение о чистой резистентности при сочетании двух препаратов, как при стандартной индукционной терапии ОМЛ [54].
Модели ксенотрансплантатов
Даже самый клинически агрессивный образец лейкозных клеток от пациента вряд ли вырастет при помещении в культуру, а характеристики клеток могут измениться в зависимости от используемой среды [55–56]. Кроме того, системы культивирования не могут воспроизводить взаимодействия между лейкозными клетками и их стромой и иммунными клетками. Чтобы обойти эти ограничения, были разработаны мышиные модели ксенотрансплантатов, позволяющие приживить первичные образцы пациентов и клеточные линии.
В первых попытках ксенотрансплантации ОМЛ мышам с ослабленным иммунитетом использовались бестимусные голые мыши, несущие мутацию в гене Foxn1 , что привело к почти полному отсутствию функциональных Т-клеток. Ранние эксперименты с использованием первичных образцов пациентов действительно привели к образованию гранулоцитарных сарком, но без вовлечения костного мозга и других органов [57]. В попытке улучшить приживление костного мозга мышей с тяжелым комбинированным иммунодефицитом (SCID) тестировали на их способность приживать образцы пациентов с AML.У этих мышей есть мутации в гене Prkdc и, следовательно, отсутствуют функциональные Т- или В-клетки. Хотя образцы, введенные внутрибрюшинно или имплантированные в капсулы почек, имели значительную скорость отбора, внутривенная инъекция привела к приживлению очень небольшого количества образцов у животных-реципиентов [58]. В важной статье доктор Джон Дик и его коллеги вводили первичные образцы пациентов мышам SCID, которых затем лечили фактором стволовых клеток человека (SCF) и фактором, стимулирующим колонии гранулоцитов-макрофагов (GM-CSF).Это привело к высокому количеству заборов периферической крови или костного мозга пациента, введенных внутривенно; что еще более важно, только часть лейкозных клеток могла успешно прижиться [10]. Это было первое свидетельство иерархии лейкозных клеток, при которой меньшинство населения отвечает за поддержание болезни. С тех пор были выведены мыши с более полным иммунодефицитом. У мышей с диабетом без ожирения (NOD) / SCID нет функциональных B- или T-клеток и снижена активность NK-клеток и моноцитов.У них более высокая скорость приживления первичных образцов пациентов, чем у мышей SCID [59]. Дальнейшее ослабление иммунитета было установлено у мышей NOD / SCID, у которых есть делеции в гене, кодирующем гамма-цепь рецептора интерлейкина 2 (IL2Rγ) (так называемые мыши NSG). Наконец, на этой платформе была создана трансгенная мышь, которая экспрессирует человеческий iL3, GM-CSF и SCF (NSG-tg). Эта мышь значительно улучшила скорость приживления первичных образцов пациентов по сравнению со стандартной мышью NSG [60]. Хотя возможности этих моделей ограничены в отношении взаимодействий лейкозных иммунных клеток, они являются ценными инструментами для оценки доклинической активности многообещающих агентов и взаимодействий лейкозных клеток со стромой.
Немышиные модели
Для изучения ОМЛ использовались другие животные модели, включая крыс. В то время как спонтанные лейкемии у крыс редки, сообщалось о многих моделях, вызванных канцерогенами и радиацией [61–64]. Миеломоноцитарный лейкоз, L5222, был индуцирован в 1967 г., через 326 дней после однократной внутривенной инъекции этилнитрозомочевины (200 мг / кг массы тела) трехмесячной самке крысы BD IX. Этот лейкоз был трансплантирован крысам BD IX и использовался в доклинических исследованиях эффективности [65].Модель APL, созданная на крысах Brown Norway (BNML), использовалась в доклинических исследованиях химиотерапии и трансплантации [66]. Лейкоз был индуцирован у самок крыс линии BN 9,10-диметил-1,2-бензантраценом [67] и имеет характеристики образования колоний, аналогичные характеристикам образования человеческих образцов AML в анализах колоний [68]. Кроме того, эта модель внесла важный вклад в понимание минимальной остаточной болезни при ОМЛ (см. Обзор [69]). Совсем недавно появились сообщения о системах AML, не относящихся к млекопитающим.Одним из примеров является недавний отчет о трансплантации образцов AML человека эмбрионам рыбок данио. В этом исследовании исследователи вводили человеческие клеточные линии AML и образцы пациентов в эмбрионы рыбок данио через 48 часов после оплодотворения. Они использовали от 50 до 200 клеток и увидели снижение лейкемической нагрузки, когда эмбрионы, которым вводили чувствительные клетки, обрабатывали иматинибом [70]. В другом исследовании человеческие APL и CML в клеточных линиях миелоидного бластного кризиса вводили эмбрионам рыбок данио; Затем эмбрионы обрабатывали иматинибом или ATRA, и у них отмечалось снижение лейкемической нагрузки [71].
Другой модельной системой, используемой для изучения AML, является плодовая муха, Drosophila melanogaster . Экспрессия слитого белка AML1-ETO в клетках крови Drosophila нарушала дифференцировку клеток крови, которые полагаются на RUNX1, подобно тому, что наблюдается у пациентов [72]. Эти системы, не относящиеся к млекопитающим, обладают преимуществами более низкой стоимости и повышенной репродуктивной способности в системах с хорошо изученной генетикой. Они могут внести значительный вклад в понимание патогенеза ОМЛ.
Модели ВСЕ на животных
ВСЕ — наиболее распространенная лейкемия у детей, а также встречается у значительного числа взрослых. Для него характерно агрессивное разрастание клональных лимфобластов. ОЛЛ имеет несколько подтипов, включая пре-В-клетки, Т-клетки и зрелые В-клетки или клетки Беркитта. Он может проявляться в основном в виде твердой массы (лимфобластные лимфомы) или с первичным поражением костного мозга (лимфобластные лейкемии). В отличие от AML, сейчас считается, что более 90% детей с диагнозом ALL могут быть излечены [73].Хотя это замечательное достижение является результатом многих направлений исследований, несколько важных открытий в отношении ALL было обнаружено с использованием моделей на животных. Эти выводы включают демонстрацию того, что активация Notch-1 приводит к Т-клеточному ОЛЛ на мышиной модели трансплантата [74], открытие, что у трансгенных мышей, экспрессирующих версию слияния BCR-ABL p190, развился ОЛЛ [75], и доклиническую оценку активность антиметаболитов [76].
Мышиные модели
Как и в случае AML, мышиные модели ALL используют канцероген-индуцированный, вирусно-индуцированный, трансгенный, мозаичный и ксенотрансплантный подходы.Эти модели внесли свой вклад в нынешнее понимание ВСЕГО.
Модели, индуцированные канцерогенами
Как и в случае AML, ранние исследования на мышах основывались на модели, индуцированной канцерогеном. В дополнение к важной клеточной линии L1210, обсужденной выше, существует несколько других канцероген-индуцированных линий ALL, включая клеточную линию L5178Y. Эта клетка является производной DBA / 2-производной метилхолантрен-индуцированной Т-клеточной линии, которая широко использовалась в доклинических исследованиях эффективности и механизмов устойчивости [77–79].
Вирусные модели
Штамм мышей AKR, склонный к лейкемии, был использован для характеристики онкогенных свойств различных вирусов. В результате была получена модель Т-клеточного ОЛЛ у мышей AKR, инфицированных рекомбинантным ретровирусом, нацеленным на тимоциты [80]. Эта модель использовалась для оценки влияния ожирения на прогрессирование ОЛЛ [81]. Кроме того, лимфобластные лейкозы и лимфомы, возникшие в результате мутагенеза вставки ретровируса, позволили охарактеризовать новые гены, участвующие в лимфоидном лейкемогенезе, путем характеристики общих последовательностей вставки вируса [82].Надежность методов была подтверждена, когда было обнаружено, что идентифицированный с помощью скрининга ген Prdm14 сверхэкспрессируется в человеческом B- и T-клеточном ОЛЛ и может инициировать ОЛЛ при сверхэкспрессии вирусной трансдукцией клеток костного мозга мыши [83].
Трансгенные модели
Было опубликовано несколько ВСЕ трансгенных моделей. Эти модели воспроизводят В-клетки, Т-клетки и лейкоз Беркитта / лимфобластные лимфомы.
BCR-ABL
Слитый белок BCR-ABL является результатом транслокации t (9; 22) (q34; q11), также известной как филадельфийская хромосома.Эта транслокация характерна для ХМЛ, но также обнаруживается при В-клеточном ОЛЛ. Существует несколько изоформ этого слитого белка, форма p210 (обнаруживается в основном при CML) и форма p190 (обнаруживается в основном при ALL). Были созданы трансгенные мыши, экспрессирующие изоформу p190 с промотора BCR, но у них была эмбриональная летальность [75]. Напротив, трансгенные мыши, экспрессирующие слияние p190 BCR-ABL с промотора металлотионеина, рождаются живыми и 95% умирают от пре-B-клеточного лейкоза / лимфомы через 35–200 дней [84].Эта модель была использована для тестирования активности новых соединений [85], ингибиторов тирозинкиназы [86] и влияния ожирения на прогрессирование ОЛЛ [81].
MLL Leukemias
Как следует из названия, лейкемия смешанного происхождения, транслокации MLL наблюдаются как при ОМЛ, так и при ОЛЛ. Слитый белок MLL-AF4 наблюдается у пациентов с t (4; 11), и он сильно связан с В-клеточным ОЛЛ. Была создана модель трансгенной мыши, которая экспрессировала MLL-AF4 с эндогенного промотора MLL. Полученные мыши продемонстрировали нерегулируемый лимфоидный и миелоидный рост, а после длительного латентного периода — В-клеточные лимфомы [87].Другая модель была создана с использованием инвертированного аллеля AF4, нацеленного на интрон в гене MLL; при воздействии рекомбиназы Cre происходила инверсия и образование MLL-AF4. Затем мышей скрещивали с различными трансгенными мышами с тканеспецифической экспрессией Cre. Когда присутствовал Cre, у мышей развивались злокачественные новообразования В-клеток с различной латентностью в зависимости от промотора Cre [88]. Трансгенная модель с использованием слитого белка MLL-AF4 человека, экспрессированного из вирусного LTR MSCV, привела к В-клеточным новообразованиям, и латентность могла быть сокращена путем скрещивания этих животных с трансгенными мышами, экспрессирующими K-ras G12D [89].Этот результат подтверждает гипотезу множественного попадания в лейкемогенез.
Eµ-myc
Лимфома / лейкемия Беркитта считается подтипом ОЛЛ и характеризуется транслокациями, в результате которых ген MYC экспрессируется под контролем промоторов тяжелой или легкой цепи иммуноглобулина. В результате болезнь характеризуется высокой скоростью распространения и очень агрессивным поведением. Были получены трансгенные мыши с мышиным геном Myc под контролем промотора тяжелой цепи IgG, имитирующего t (8; 14), наблюдаемый в клинических условиях.В результате у мышей развились агрессивные В-клеточные лимфомы и лейкозы, при этом 90% животных умерли в течение первых 5 месяцев жизни [90]. Эта модель была использована для изучения роли взаимодействующих мутаций в лимфоме / лейкемогенезе [91], а также в основополагающей статье о том, как потеря p53 или избыточная экспрессия BCL2 влияет на химиотерапевтический ответ in vivo [92].
NOTCh2
Рецепторы Notch — это трансмембранные рецепторы, которые при активации лигандом расщепляются на внеклеточные и внутриклеточные части.Внутриклеточная часть (ICN) перемещается в ядро, где она взаимодействует с дополнительными белками, что приводит к транскрипционной активации генов-мишеней (обзор в [93]). NOTCh2 был впервые идентифицирован при транслокации, несущей ALL, Т-лимфоциты человека, t (7; 9) (q34; q34.3). Эта транслокация приводит к образованию гена слияния, в котором промотор TCR управляет экспрессией 3 ’конца локуса NOTCh2 , кодируя только ICN и приводя к конститутивной активации NOTCh2 [94].Эта транслокация обнаруживается только примерно в 1% Т-клеточного ОЛЛ. Однако в последующем сообщении от 50 до 60% образцов Т-клеточного ОЛЛ содержали точечные мутации в NOTCh2, , причастные к патогенезу большинства типов Т-клеточного ОЛЛ [95]. Было создано несколько моделей трансгенных мышей для экспрессии активированных аллелей NOTCh2 из Т-клеточных промоторов [96–98]. У этих мышей изменено развитие тимоцитов CD4, CD8 и развиваются злокачественные опухоли Т-клеток. В более поздней модели используется трансгенная мышь, которая условно экспрессирует две общие точечные мутации в NOTCh2 из своего эндогенного промотора с использованием кассеты Lox-Stop-Lox.Эти мыши продемонстрировали ускоренное развитие злокачественных опухолей Т-клеток в системе мутагенеза транспозонов Спящей красавицы [99].
Mosaic Models
Как и в случае AML, ex-vivo манипуляции с HSC были использованы для создания мышиных моделей ALL. MSCV-векторы, сконструированные для экспрессии слитого белка p190 BCR-ABL, широко использовались для создания моделей ALL B-клеток, положительных по филадельфийской хромосоме. Несколько примеров включают исследование эффектов химиотерапии на развитие мутантов BCR-ABL, устойчивых к ингибиторам тирозинкиназы (TKI) [100], роль BCL6 в устойчивости к TKI [101] и роль супрессора опухолей Arf. в развитии В-клеточного ОЛЛ [102].Онкогены слияния MLL также широко использовались для создания мозаичных ВСЕХ моделей. Исследователи объединили мозаичную модель с моделью ксенотрансплантата и, используя человеческие HSC, полученные из пуповинной крови, инфицировали их вектором, экспрессирующим MLL-ENL на основе MSCV, и вводили их сублетально облученным мышам NOD / SCID. У 26 из 29 мышей, которым вводили инъекцию, развился агрессивный В-клеточный ОЛЛ, демонстрирующий, что MLL-ЭНЛ может индуцировать ОЛЛ в клетках человека [103]. В дополнительном исследовании слияние MLL-AF9 могло продуцировать AML или ALL в CD34 + клетках пуповинной крови человека в зависимости от мыши-реципиента (NSG против NSG-tg) [104].Эти исследования установили возможность создания моделей ОЛЛ из человеческих клеток, размножающихся у мышей с ослабленным иммунитетом, и начали разгадывать, каким образом лейкозы с перегруппировкой MLL могут быть миелоидными или лимфоидными.
Модели ксенотрансплантатов
Те же линии мышей с ослабленным иммунитетом, описанные в разделе ксенотрансплантатов в моделях AML, были использованы для проведения доклинических исследований эффективности во ВСЕХ образцах. Кроме того, они распространили модель лейкозных стволовых клеток на ОЛЛ, наблюдая, что не все В- или Т-клеточные ОЛЛ-клетки могут инициировать лейкемию у мышей NOD / SCID или NSG [105–106].
Немышиные модели
Подразделение инбредных крыс Sprague-Dawley имеет высокую частоту Т-клеточных злокачественных новообразований и использовалось в доклинической оценке терапевтических средств [107–108]. В-клеточный ОЛЛ был получен от инбредных морских свинок и был серийно трансплантирован инбредным реципиентам, но не беспородным животным [109]. Существуют также модели ОЛЛ, не относящиеся к млекопитающим, включая трансгенную модель рыбок данио, основанную на экспрессии слитого белка TELAML1, обнаруженного у пациентов с пре-В-клеточным ОЛЛ с t (12; 21) (p13; q22).Исследователи экспрессировали трансген либо повсеместно (с использованием модифицированного промотора актина), либо ограниченным лимфоидом (с использованием промотора Rag2). Только у рыбок данио, которые экспрессировали трансген повсеместно, развивался ОЛЛ после длительного латентного периода [110]. Скрининг химического мутагенеза с использованием трансгенных рыбок данио, которые экспрессируют усиленный зеленый флуоресцентный белок Т-клеточно-специфическим образом, дал несколько линий, которые имеют наследственный Т-клеточный ОЛЛ [111]. Другая модель ОЛЛ Т-клеток была создана у рыбок данио с использованием мышиного гена c-myc , экспрессируемого под контролем промотора Rag2 рыбок данио.У трансгенных рыб развился Т-клеточный ОЛЛ с короткой латентностью [112]. В элегантной модели доктор Лук и его коллеги использовали трансгенных рыбок данио, которые содержали условный аллель Cre, управляемый промотором теплового шока 70, и скрестили его с трансгенной моделью, содержащей мышиный c-myc , управляемый промотором Rag2 , содержащим промежуточная кассета Lox-stop-DS Red-Lox. Полученное потомство можно было поместить в воду при 37 ° C, чтобы вызвать экспрессию Cre. Иссечение стоп-кассеты может сопровождаться флуоресцентной визуализацией для обнаружения потери DS Red и индукции экспрессии EGFP.У этих животных развилась Т-клеточная лимфобластная лимфома (LBL), экспрессирующая EGFP, которая после лечения тепловым шоком прогрессировала до лейкемии [113]. Эта модель позже была использована для исследования генетических событий, которые приводят к превращению LBL в ALL [114]. У рыбок данио хорошо зарекомендовавшая себя генетика, низкая стоимость и высокие показатели воспроизводства, что делает их идеальной системой для проведения генетических скринингов. Вклад этих моделей только начинается.
Животные модели CML
ХМЛ уникален среди лейкозов тем, что существует единственное онкогенное событие, которое вызывает заболевание — слитый белок BCR-ABL, генерируемый филадельфийской хромосомой.Это была первая известная соматическая хромосомная аномалия, напрямую связанная со злокачественным новообразованием, и стало первым доказательством того, что рак является генетическим заболеванием [115]. Заболевание характеризуется аномальной пролиферацией и накоплением зрелых и созревающих миелоидных клеток. Он имеет три различных клинических фазы: хроническую фазу, ускоренную фазу и фазу взрыва. В прошлом ХМЛ можно было вылечить только трансплантацией аллогенных стволовых клеток, но в последние годы лечение произвело революцию с появлением ингибиторов киназы BCR-ABL, таких как иматиниб, нилотиниб и дазатиниб (обзор в [116]).
Животные модели сыграли уникальную роль в исследованиях ХМЛ. Доктор Ван Эттен показал, что изоформа p210 белка BCR-ABL непосредственно вызывает ХМЛ, используя мозаичную модель мыши. Он использовал ретровирусный вектор, который экспрессировал белок p210 и инфицированные мышиные HSC. Когда инфицированные клетки были трансплантированы сингенным реципиентам, у животных развилось миелопролиферативное заболевание, сильно напоминающее ХМЛ человека [117]. С тех пор эти модели широко использовались для оценки многих аспектов ХМЛ, включая трансформирующий потенциал различных изоформ BCR-ABL [118], генетику прогрессирования заболевания [36] и характеристику стволовых клеток ХМЛ [119]. .
CML также моделировали с использованием трансгенных мышей. В одной модели изоформа p210 экспрессировалась из индуцируемого тетрациклином протомера и скрещивалась с трансгенной мышью, которая экспрессировала tet-трансактиватор с промотора CD34, обеспечивая экспрессию p210 в присутствии доксициклина в компартменте HSC. В результате у мышей развилось миелопролиферативное заболевание с тромбоцитозом [120]. Это противоположно B-клеточному ALL, которое они наблюдали, когда p210 экспрессировался в пре-B-клетках [121], демонстрируя, что фенотип лейкозов, индуцированных BCR-ABL, зависит, по крайней мере, частично от клетки-источника.В дополнение к мозаичным и трансгенным моделям были проведены исследования ксенотрансплантатов с использованием образцов пациентов с ХМЛ. В недавнем примере сохранение популяции стволовых клеток ХМЛ у пациентов в цитогенетической ремиссии было продемонстрировано путем приживления клеток мышам NSG [122]. Это исследование доказывает устойчивость этих лейкозных стволовых клеток даже у пациентов, которые получали терапию TKI в течение многих лет. Первичные образцы пациентов и клеточные линии, полученные от пациентов с ХМЛ в различных фазах, были приживлены бестимусным мышам [123], мышам SCID [124] и мышам NOD / SCID [125].Использование мозаичных, трансгенных и ксенотрансплантных моделей дало неоценимую информацию о молекулярной патологии этого лейкоза.
Животные модели ХЛЛ
ХЛЛ является наиболее распространенной из всех лейкозов и, в отличие от ХМЛ, является генетически гетерогенным заболеванием. Это заболевание пожилых людей, характеризующееся клональной пролиферацией иммунодефицитных CD5 + зрелых В-лимфоцитов. Заболевание считается вялотекущим, при этом средняя выживаемость при низкой стадии заболевания составляет более десяти лет, хотя у подгруппы пациентов будет более скоротечное течение [126].Заболевание может иногда трансформироваться в более агрессивную диффузную крупноклеточную В-клеточную лимфому или, реже, в лимфому Ходжкина посредством процесса, называемого трансформацией Рихтера. К настоящему времени описано несколько моделей ХЛЛ на животных. Как и в случае с другими лейкозами, мыши являются наиболее широко используемой моделью.
Одной из самых ранних моделей мышей с ХЛЛ является спонтанно возникшая модель мышей новозеландских черных (NZB). У мышей NZB развивается клональная пролиферация анеуплоидных лимфоцитов CD5 + B. Заболевание может быть пересажено серийно и иногда превращается в крупноклеточную лимфому, соответствующую трансформации Рихтера, наблюдаемой клинически [127].У этих мышей была обнаружена мутация в локусе микроРНК-16; также наблюдаемый в клетках ХЛЛ человека, считается, что он способствует развитию ХЛЛ [128]. В дополнение к этой спонтанной модели было разработано несколько трансгенных моделей. В трансгенной модели TCL1 человеческий ген TCL1 был клонирован ниже промотора E, что привело к ограничению экспрессии B-клеток на высоком уровне. Ген TCL1 имеет повышенную экспрессию при ряде злокачественных новообразований В-клеток, включая ХЛЛ [129].Полученные мыши развивают клональную или олигоклональную пролиферацию лимфоцитов CD5 + B в более позднем возрасте, имитируя болезнь человека [130]. Дополнительное понимание молекулярной патологии ХЛЛ было получено с помощью двойной трансгенной модели TRAF2DN / Bcl-2. Как Bcl-2, так и факторы, ассоциированные с рецептором фактора некроза опухоли (TRAF), сверхэкспрессируются при злокачественных новообразованиях B-клеток, включая CLL [131–132]. В этой модели исследователи скрестили трансгенных животных, экспрессирующих человеческий BCL2 с промотора тяжелой цепи IgG, с животными, экспрессирующими усеченную форму TRAF2.Полученное потомство развивало прогрессивное накопление клональных CD-экспрессирующих В-клеток, что приводило к сокращению выживаемости [133]. Как и в случае с другими лейкозами, сообщалось о распространении образцов ХЛЛ человека у мышей с ослабленным иммунитетом. Полученная от человека линия клеток CLL, устойчивых к флударабину, WSUCLL, может вызывать опухоли при подкожном введении мышам SCID [134]. Эта модель использовалась для доклинических испытаний [135]. Голые мыши также служили реципиентами клеточных линий CLL человека [136]. Наконец, животных NOD / SCID использовали для приживления первичных образцов пациентов [137].Эта модель была использована для изучения эффектов известных прогностических маркеров ХЛЛ на приживление образца; образцы с неблагоприятными факторами прививаются более агрессивно в этой модели [138]. Первые успехи этой модели делают ее очень привлекательной для будущих доклинических исследований эффективности новых агентов.
Резюме и направления на будущее
Животные модели лейкемии были незаменимы в изучении этих разрушительных болезней. Можно привести убедительный аргумент в пользу того, что наше понимание лейкозов больше, чем любое другое злокачественное новообразование, выиграло от этих модельных систем.Большая часть нынешнего понимания и терапевтических подходов были впервые проверены или открыты с использованием этих моделей. В будущем можно легко представить синергию между различными моделями для дальнейшей разработки новых терапевтических целей. Трансгенные и мозаичные модели имеют очевидную полезность для доклинических испытаний эффективности, а также для изучения взаимодействий стромы и иммунных клеток. У моделей ксенотрансплантатов есть уникальное преимущество, заключающееся в том, что они могут исследовать специфическую биологию клеток человека в in vivo, хотя и в среде с ослабленным иммунитетом.Новые системы, не относящиеся к млекопитающим, могут привести к крупномасштабному генетическому скринингу для выявления критических молекулярных путей, которые могут служить следующим поколением терапевтических мишеней, которые будут оценены на мозаичных, трансгенных и ксенотрансплантных моделях и переведены в клинические испытания для улучшения жизни. наших пациентов с лейкемиями.
Благодарности
Авторы выражают благодарность Карен Кляйн за помощь в редактировании рукописи. Поддержка предоставлена Фондом Дуга Коли по исследованиям лейкемии, Фондом Маккея по исследованиям рака, Фондом исследований Лейта и Фондом Фрэнсис П. Тутуилер.
Библиография
1. Сигел Р., Уорд Э, Броули О., Джемаль А. Статистика рака, 2011: Влияние устранения социально-экономических и расовых различий на преждевременную смерть от рака. CA Cancer J Clin. 2011. 61: 212–236. [PubMed] [Google Scholar] 2. Гиллиланд Д.Г., Джордан Коннектикут, Феликс Калифорния. Молекулярные основы лейкемии. Образовательная программа Hematology Am Soc Hematol. 2004: 80–97. [PubMed] [Google Scholar] 3. Licht JD, Sternberg DW. Молекулярная патология острого миелолейкоза. Образовательная программа Hematology Am Soc Hematol.2005: 137–142. [PubMed] [Google Scholar] 4. Ловенберг Б. Острый миелоидный лейкоз: проблема выявления разнообразия болезней. Образовательная программа Hematology Am Soc Hematol. 2008; 2008: 1–11. [PubMed] [Google Scholar] 5. Dohner H, Estey EH, Amadori S и др. Диагностика и лечение острого миелоидного лейкоза у взрослых: рекомендации международной группы экспертов от имени European Leukemia Net. Кровь. 2010. 115: 453–474. [PubMed] [Google Scholar] 6. Нерви Б., Рамирес П., Реттиг М.П. и др. Хемосенсибилизация острого миелоидного лейкоза (ОМЛ) после мобилизации антагонистом CXCR4 AMD3100.Кровь. 2009. 113: 6206–6214. [Бесплатная статья PMC] [PubMed] [Google Scholar] 7. Jaiswal S, Jamieson CH, Pang WW, et al. CD47 активируется циркулирующими гемопоэтическими стволовыми клетками и лейкозными клетками, чтобы избежать фагоцитоза. Клетка. 2009. 138: 271–285. [Бесплатная статья PMC] [PubMed] [Google Scholar] 8. Маджети Р., Чао М.П., Ализаде А.А. и др. CD47 является неблагоприятным прогностическим фактором и мишенью терапевтических антител для стволовых клеток острого миелоидного лейкоза человека. Клетка. 2009. 138: 286–299. [Бесплатная статья PMC] [PubMed] [Google Scholar] 9.Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Лейкозные клетки создают ниши костного мозга, которые нарушают поведение нормальных кроветворных клеток-предшественников. Наука. 2008; 322: 1861–1865. [PubMed] [Google Scholar] 10. Лапидот Т., Сирард С., Формор Дж. И др. Клетка, инициирующая острый миелоидный лейкоз человека после трансплантации мышам SCID. Природа. 1994; 367: 645–648. [PubMed] [Google Scholar] 11. Джордан CT, Гусман ML, Noble M. Раковые стволовые клетки. N Engl J Med. 2006; 355: 1253–1261. [PubMed] [Google Scholar] 12.Skipper HE, Perry S. Кинетика нормальных и лейкозных популяций лейкоцитов и отношение к химиотерапии. Cancer Res. 1970; 30: 1883–1897. [PubMed] [Google Scholar] 13. Закон LW, Таормина V, Бойл PJ. Ответ острых лимфолейкозов на антагонист пуринов 6-меркаптопурин. Ann N Y Acad Sci. 1954. 60: 244–250. [PubMed] [Google Scholar] 14. Маккормак Э., Брюзруд О., Гьертсен БТ. Животные модели острого миелогенного лейкоза — разработка, применение и перспективы на будущее. Лейкемия. 2005; 19: 687–706.[PubMed] [Google Scholar] 15. Шкипер HE, Schabel FM, Jr, Wilcox WS. Экспериментальная оценка потенциальных противораковых агентов. XXI. Планирование приема арабинозилцитозина для использования его специфичности в S-фазе против лейкозных клеток. Cancer Chemother Rep. 1967; 51: 125–165. [PubMed] [Google Scholar] 16. Френд С. Бесклеточная передача у взрослых швейцарских мышей болезни, имеющей характер лейкемии. J Exp Med. 1957; 105: 307–318. [Бесплатная статья PMC] [PubMed] [Google Scholar] 17. Линемейер Д.Л., Менке Дж. Г., Рускетти С. К., Эванс Л. Х., Сколник Е. М..Последовательности гена оболочки, которые кодируют белок gp52 фокусообразующего вируса селезенки, необходимы для индукции пролиферации эритроидных клеток. J Virol. 1982; 43: 223–233. [Бесплатная статья PMC] [PubMed] [Google Scholar] 18. Вольф Л., Рускетти С. Злокачественная трансформация эритроидных клеток in vivo путем введения нереплицирующегося ретровирусного вектора. Наука. 1985; 228: 1549–1552. [PubMed] [Google Scholar] 19. Back J, Dierich A, Bronn C, Kastner P, Chan S. PU.1 определяет способность эритроидных клеток-предшественников к самообновлению.Кровь. 2004. 103: 3615–3623. [PubMed] [Google Scholar] 20. Erkeland SJ, Valkhof M, Heijmans-Antonissen C, et al. Масштабная идентификация генов болезней, вызывающих острый миелоидный лейкоз. J Virol. 2004; 78: 1971–1980. [Бесплатная статья PMC] [PubMed] [Google Scholar] 21. Кауделл Д., Харпер Д.П., Новак Р.Л. и др. Ретровирусный инсерционный мутагенез идентифицирует активацию Zeb2 как новое лейкемогенное событие взаимодействия у трансгенных мышей CALM-AF10. Кровь. 2010; 115: 1194–1203. [Бесплатная статья PMC] [PubMed] [Google Scholar] 22.Slape C, Hartung H, Lin YW, Bies J, Wolff L, Aplan PD. Ретровирусный инсерционный мутагенез идентифицирует гены, которые взаимодействуют с NUP98-HOXD13 во время лейкемической трансформации. Cancer Res. 2007. 67: 5148–5155. [Бесплатная статья PMC] [PubMed] [Google Scholar] 23. Шкипер HE, Schabel FM, младший, трейдер MW, Laster WR., Младший Ответ на терапию спонтанного, первого прохождения и длинных линий прохождения лейкемии AK. Cancer Chemother Rep., 1969; 53: 345–366. [PubMed] [Google Scholar] 24. Василиу Г.С., Купер Дж. Л., Рад Р. и др.Мутантный нуклеофозмин и взаимодействующие пути приводят к возникновению и прогрессированию лейкемии у мышей. Нат Жене. 2011; 43: 470–475. [Бесплатная статья PMC] [PubMed] [Google Scholar] 25. Коллиер Л.С., Адамс Д.Д., Хакетт С.С. и др. Мутагенез спящей красавицы всего тела может вызвать пенетрантную лейкемию / лимфому и редкую глиому высокой степени злокачественности без связанной с этим эмбриональной летальности. Cancer Res. 2009; 69: 8429–8437. [Бесплатная статья PMC] [PubMed] [Google Scholar] 26. Heyer J, Kwong LN, Lowe SW, Chin L. Генно-инженерные модели мышей, не являющиеся зародышевыми линиями, для трансляционных исследований рака.Нат Рев Рак. 2010; 10: 470–480. [Бесплатная статья PMC] [PubMed] [Google Scholar] 27. Early E, Moore MA, Kakizuka A, et al. Трансгенная экспрессия PML / RARalpha нарушает миелопоэз. Proc Natl Acad Sci U S. A. 1996; 93: 7900–7904. [Бесплатная статья PMC] [PubMed] [Google Scholar] 28. Браун Д., Коган С., Лагассе Е. и др. Трансген PMLRARalpha вызывает острый промиелоцитарный лейкоз у мышей. Proc Natl Acad Sci U S. A. 1997; 94: 2551–2556. [Бесплатная статья PMC] [PubMed] [Google Scholar] 29. Гризолано Дж. Л., Весселшмидт Р. Л., Пеличчи П. Г., Лей Т. Дж..Измененное миелоидное развитие и острый лейкоз у трансгенных мышей, экспрессирующих PML-RAR альфа под контролем регуляторных последовательностей катепсина G. Кровь. 1997. 89: 376–387. [PubMed] [Google Scholar] 30. He LZ, Guidez F, Tribioli C и др. Различное взаимодействие PML-RARalpha и PLZF-RARalpha с корепрессорами определяет дифференциальные ответы на RA в APL. Нат Жене. 1998. 18: 126–135. [PubMed] [Google Scholar] 31. Зубер Дж., Радтке И., Парди Т.С. и др. Мышиные модели ОМЛ человека точно предсказывают ответ на химиотерапию.Genes Dev. 2009; 23: 877–889. [Бесплатная статья PMC] [PubMed] [Google Scholar] 32. Гримуэйд Д., Уокер Х., Оливер Ф. и др. Важность диагностической цитогенетики для исходов при ОМЛ: анализ 1612 пациентов, включенных в исследование MRC AML-10. Рабочие группы Совета медицинских исследований по лейкемии взрослых и детей. Кровь. 1998. 92: 2322–2333. [PubMed] [Google Scholar] 33. Окуда Т., Цай З., Ян С. и др. Экспрессия заблокированного гена лейкемии AML1-ETO ингибирует установление нормального дефинитивного гематопоэза и непосредственно генерирует диспластические гематопоэтические предшественники.Кровь. 1998. 91: 3134–3143. [PubMed] [Google Scholar] 34. Роадс К.Л., Хетерингтон С.Дж., Харакава Н. и др. Анализ роли AML1-ETO в лейкемогенезе с использованием модели индуцибельных трансгенных мышей. Кровь. 2000; 96: 2108–2115. [PubMed] [Google Scholar] 35. Юань Ю., Чжоу Л., Миямото Т. и др. Экспрессия AML1-ETO напрямую участвует в развитии острого миелоидного лейкоза при наличии дополнительных мутаций. Proc Natl Acad Sci U S. A. 2001; 98: 10398–10403. [Бесплатная статья PMC] [PubMed] [Google Scholar] 36.Даш А.Б., Уильямс И.Р., Куток Дж. Л. и др. Мышиная модель бластного кризиса ХМЛ, вызванного сотрудничеством между BCR / ABL и NUP98 / HOXA9. Proc Natl Acad Sci U S. A. 2002; 99: 7622–7627. [Бесплатная статья PMC] [PubMed] [Google Scholar] 37. Гризолано Дж. Л., О’Нил Дж., Каин Дж., Томассон М. Х. Активированная рецепторная тирозинкиназа, TEL / PDGFbetaR, взаимодействует с AML1 / ETO, вызывая острый миелоидный лейкоз у мышей. Proc Natl Acad Sci U S. A. 2003; 100: 9506–9511. [Бесплатная статья PMC] [PubMed] [Google Scholar] 38. Lavau C, Du C, Thirman M, Zeleznik-Le N.Связанные с хроматином свойства CBP, слитого с MLL, вызывают миелодиспластический синдром, который перерастает в миелоидный лейкоз. EMBO J. 2000; 19: 4655–4664. [Бесплатная статья PMC] [PubMed] [Google Scholar] 39. Смотреть на. Онкогенные факторы транскрипции при острых лейкозах человека. Наука. 1997; 278: 1059–1064. [PubMed] [Google Scholar] 40. Daser A, Rabbitts TH. Расширение репертуара гена лейкемии смешанного происхождения MLL в лейкемогенезе. Genes Dev. 2004; 18: 965–974. [PubMed] [Google Scholar] 41. Corral J, Lavenir I, Impey H и др.Ген слияния Mll-AF9, полученный путем гомологичной рекомбинации, вызывает острый лейкоз у химерных мышей: метод создания слитых онкогенов. Клетка. 1996. 85: 853–861. [PubMed] [Google Scholar] 42. Стриссель П.Л., Стрик Р., Томек Р.Дж., Роу Б.А., Роули Д.Д., Железник-Ле, штат Нью-Джерси. Структурные свойства ДНК AF9 аналогичны MLL и могут действовать как горячие точки рекомбинации, приводящие к транслокациям MLL / AF9 и лейкемогенезу. Hum Mol Genet. 2000; 9: 1671–1679. [PubMed] [Google Scholar] 43. Коллинз Э.С., Паннелл Р., Симпсон Э.М., Форстер А., Кролик Т.Х.Межхромосомная рекомбинация генов Mll и Af9, опосредованная cre-loxP в развитии мышей. EMBO Rep. 2000; 1: 127–132. [Бесплатная статья PMC] [PubMed] [Google Scholar] 44. Форстер А., Паннелл Р., Дринан Л. Ф. и др. Разработка de novo реципрокных хромосомных транслокаций, связанных с Mll, для репликации первичных событий рака человека. Раковая клетка. 2003. 3: 449–458. [PubMed] [Google Scholar] 45. Стируолт Д.Л., Радич Ю.П. Роль FLT3 в злокачественных заболеваниях кроветворения. Нат Рев Рак. 2003. 3: 650–665. [PubMed] [Google Scholar] 46.Ли Л., Пилото О., Нгуен Х. Б. и др. Включение внутренней тандемной дупликационной мутации в мышиный FLT3 вызывает миелопролиферативное заболевание на мышиной модели. Кровь. 2008; 111: 3849–3858. [Бесплатная статья PMC] [PubMed] [Google Scholar] 47. Гринблатт С., Ли Л., Слейп С и др. Включение мутации FLT3 / ITD кооперируется со слиянием NUP98-HOXD13 для генерации острого миелоидного лейкоза на модели мышей. Кровь. 2012 [Бесплатная статья PMC] [PubMed] [Google Scholar] 48. Чан ИТ, Гиллиланд Д.Г. Онкогенные K-ras в мышиных моделях миелопролиферативного заболевания и острого миелоидного лейкоза.Клеточный цикл. 2004; 3: 536–537. [PubMed] [Google Scholar] 49. Маккензи К.Л., Дольников А., Миллингтон М., Шоунан Ю., Симондс Г. Мутантные N-расы вызывают миелопролиферативные нарушения и апоптоз у мышей с репопуляцией костного мозга. Кровь. 1999; 93: 2043–2056. [PubMed] [Google Scholar] 50. Мартелли AM, Nyakern M, Tabellini G, et al. Сигнальный путь фосфоинозитид-3-киназы / Akt и его терапевтические последствия для острого миелоидного лейкоза человека. Лейкемия. 2006; 20: 911–928. [PubMed] [Google Scholar] 51. Ю Х, Ли Й, Гао С. и др.Соответствующая мышиная модель моноцитарного лейкоза человека посредством Cre / lox-контролируемой миелоид-специфической делеции PTEN. Лейкемия. 2010; 24: 1077–1080. [Бесплатная статья PMC] [PubMed] [Google Scholar] 52. Каттс Б.А., Шегрен А.К., Андерссон К.М. и др. Дефицит Nf1 взаимодействует с онкогенным K-RAS, вызывая у мышей острый миелоидный лейкоз. Кровь. 2009. 114: 3629–3632. [PubMed] [Google Scholar] 53. de Guzman CG, Warren AJ, Zhang Z, et al. Экспансия гемопоэтических стволовых клеток и отчетливые аномалии развития миелоидов на мышиной модели транслокации AML1-ETO.Mol Cell Biol. 2002; 22: 5506–5517. [Бесплатная статья PMC] [PubMed] [Google Scholar] 54. Парди Т.С., Зубер Дж., Лоу SW. Flt3-ITD изменяет химиотерапевтический ответ in vitro и in vivo p53-зависимым образом. Exp Hematol. 2011; 39: 473–485. e474. [Бесплатная статья PMC] [PubMed] [Google Scholar] 55. Bruserud O, Tore Gjertsen B, Brustugun OT и др. Влияние интерлейкина 10 на бластные клетки, полученные от пациентов с острым миелогенным лейкозом. Лейкемия. 1995; 9: 1910–1920. [PubMed] [Google Scholar] 56. Bruserud O, Gjertsen BT, von Volkman HL.Культура in vitro клеток острого миелогенного лейкоза человека (AML) в бессывороточной среде: исследования нативных бластов AML и клеточных линий AML. J Hematother Stem Cell Res. 2000; 9: 923–932. [PubMed] [Google Scholar] 57. Нара Н., Миямото Т. Прямая и серийная трансплантация острого миелоидного лейкоза человека голым мышам. Br J Рак. 1982; 45: 778–782. [Бесплатная статья PMC] [PubMed] [Google Scholar] 58. Sawyers CL, Gishizky ML, Quan S, Golde DW, Witte ON. Распространение бластных миелоидных лейкозов человека у мышей SCID.Кровь. 1992; 79: 2089–2098. [PubMed] [Google Scholar] 59. Ailles LE, Gerhard B, Kawagoe H, Hogge DE. Характеристики роста предшественников острого миелогенного лейкоза, которые инициируют злокачественное кроветворение у мышей, не страдающих ожирением, диабетом / тяжелым комбинированным иммунодефицитом. Кровь. 1999; 94: 1761–1772. [PubMed] [Google Scholar] 60. Вундерлих М., Чоу Ф.С., Линк К.А. и др. Эффективность ксенотрансплантата AML значительно повышается у мышей NOD / SCID-IL2RG, конститутивно экспрессирующих SCF, GM-CSF и IL-3 человека. Лейкемия. 2010; 24: 1785–1788.[Бесплатная статья PMC] [PubMed] [Google Scholar] 61. Svejda J, Kossey P, Hlavayova E, Svec F. Гистологическая картина лейкемии трансплантируемых крыс, вызванная рентгеновским облучением и метилхолантреном. Новообразования. 1958; 5: 123–131. [PubMed] [Google Scholar] 62. Мориучи Т., Оикава Т., Кодама Т., Ямагути Х., Кобаяси Х. Создание и характеристика трансплантируемого миеломоноцитарного лейкоза крыс. Cancer Res. 1983; 43: 5478–5483. [PubMed] [Google Scholar] 63. Иванкович С., Целлер В.Дж. [Лейкемия L 5222 штамма крыс BD IX.Индуцированная этилнитрозомочевиной моноцитарно-миелоидная трансплантируемая форма для цитохимических и химиотерапевтических исследований] Блют. 1974. 28: 288–292. [PubMed] [Google Scholar] 64. Пирсон Дж. У., Чапарас С. Д., Торгерсен Дж. А., Перк К., Чиригос М. А., Шер Н. А.. Эффект лекарственной терапии против гистологически определенного лейкоза крыс. Cancer Res. 1974; 34: 355–361. [PubMed] [Google Scholar] 65. Zeller WJ, Ivankovic S, Schmahl D. Химиотерапия трансплантируемого острого лейкоза L5222 у крыс. Cancer Res. 1975. 35: 1168–1174. [PubMed] [Google Scholar] 66.Hagenbeek A, Martens AC. Эффективность высоких доз циклофосфамида в сочетании с облучением всего тела при лечении острого миелоцитарного лейкоза: исследования на соответствующей модели крыс. Cancer Res. 1983; 43: 408–412. [PubMed] [Google Scholar] 67. Hagenbeek A, Martens AC. Патогенез крысиной модели острого миелоцитарного лейкоза человека. Haematologica. 1980; 65: 293–308. [PubMed] [Google Scholar] 68. ван Беккум Д.В., ван Остером П., Дике К.А. Образование колоний in vitro при трансплантируемых лейкозах крыс по сравнению с острым миелоидным лейкозом человека.Cancer Res. 1976; 36: 941–946. [PubMed] [Google Scholar] 69. Martens AC, van Bekkum DW, Hagenbeek A. Минимальная остаточная болезнь при лейкемии: исследования на животной модели острого миелоцитарного лейкоза (BNML) Int J Cell Cloning. 1990; 8: 27–38. [PubMed] [Google Scholar] 70. Пруво Б., Жакель А., Дроин Н. и др. Ксенотрансплантат лейкозных клеток в эмбрионе рыбок данио для исследования эффективности лекарств. Haematologica. 2011; 96: 612–616. [Бесплатная статья PMC] [PubMed] [Google Scholar] 71. Коркери Д.П., Деллер Г., Берман Дж. Ксенотрансплантация лейкемии в анализе реакции на химиотерапию рыбок данио in vivo.Br J Haematol. 2011; 153: 786–789. [PubMed] [Google Scholar] 72. Osman D, Gobert V, Ponthan F, Heidenreich O, Haenlin M, Waltzer L. Модель дрозофилы идентифицирует кальпаины как модуляторы человеческого лейкемогенного слитого белка AML1-ETO. Proc Natl Acad Sci U S. A. 2009; 106: 12043–12048. [Бесплатная статья PMC] [PubMed] [Google Scholar] 73. Hunger SP, Lu X, Devidas M и др. Улучшение выживаемости детей и подростков с острым лимфобластным лейкозом в период с 1990 по 2005 год: отчет группы детской онкологии.J Clin Oncol. 2012 [Бесплатная статья PMC] [PubMed] [Google Scholar] 74. Груша В.С., Астер Дж. К., Скотт М.Л. и др. Исключительное развитие Т-клеточных новообразований у мышей с трансплантированным костным мозгом, экспрессирующим активированные аллели Notch. J Exp Med. 1996; 183: 2283–2291. [Бесплатная статья PMC] [PubMed] [Google Scholar] 75. Heisterkamp N, Jenster G, ten Hoeve J, Zovich D, Pattengale PK, Groffen J. Острый лейкоз у трансгенных мышей bcr / abl. Природа. 1990; 344: 251–253. [PubMed] [Google Scholar] 76. Law LW, Dunn TB и др. Наблюдения за действием антагониста фолиевой кислоты на трансплантируемые лимфоидные лейкозы у мышей.J Natl Cancer Inst. 1949; 10: 179–192. [PubMed] [Google Scholar] 77. Тренер DL, Уилок Э.Ф. Характеристика фенотипов клеток L5178Y, выделенных во время прогрессирования состояния покоя опухоли у мышей DBA2. Cancer Res. 1984; 44: 2897–2906. [PubMed] [Google Scholar] 78. Нишимура Т., Муто К., Танака Н. Лекарственная чувствительность устойчивой к адриамицину мутантной сублинии клеток лимфобластомы мыши L5178Y. Дж. Антибиотик (Токио) 1978; 31: 493–495. [PubMed] [Google Scholar] 79. Нисимура Т., Сузуки Х., Муто К., Танака Н. Механизм устойчивости к адриамицину в подлинии клеток лимфобластомы мыши L5178Y.Журнал «Антибиотик» (Токио), 1979; 32: 518–522. [PubMed] [Google Scholar] 80. Клойд М.В., Хартли Дж. В., Роу В.П. Лимфомагенность рекомбинантных вирусов мышиного лейкоза, индуцирующих очаговые клетки норки. J Exp Med. 1980; 151: 542–552. [Бесплатная статья PMC] [PubMed] [Google Scholar] 81. Юн Дж. П., Бехан Дж. В., Хейстеркамп Н. и др. Ожирение, вызванное диетой, ускоряет прогрессирование острого лимфобластного лейкоза в двух моделях на мышах. Cancer Prev Res (Phila) 2010; 3: 1259–1264. [Бесплатная статья PMC] [PubMed] [Google Scholar] 82. Weiser KC, Liu B, Hansen GM и др.Ретровирусные вставки в базе данных VISION идентифицируют молекулярные пути лимфолейкоза и лимфомы мышей. Мамм Геном. 2007. 18: 709–722. [Бесплатная статья PMC] [PubMed] [Google Scholar] 83. Деттман Э.Дж., Симко С.Дж., Аянга Б. и др. Prdm14 инициирует лимфобластный лейкоз после увеличения популяции клеток, напоминающих обычные лимфоидные предшественники. Онкоген. 2011; 30: 2859–2873. [Бесплатная статья PMC] [PubMed] [Google Scholar] 84. Groffen J, Voncken JW, Kaartinen V, Morris C, Heisterkamp N. Ph-позитивный лейкоз: модель трансгенных мышей.Лимфома лейка. 1993; 11 (Приложение 1): 19–24. [PubMed] [Google Scholar] 85. Reichert A, Heisterkamp N, Daley GQ, Groffen J. Лечение Bcr / Abl-положительного острого лимфобластного лейкоза у трансгенных мышей P190 с помощью ингибитора фарнезилтрансферазы SCH66336. Кровь. 2001; 97: 1399–1403. [PubMed] [Google Scholar] 86. Каур П., Фельдхан Н., Чжан Б. и др. Лечение нилотинибом на мышиных моделях лимфобластного лейкоза P190 Bcr / Abl. Молочный рак. 2007; 6: 67. [Бесплатная статья PMC] [PubMed] [Google Scholar] 87. Чен В., Ли К., Хадсон В. А., Кумар А., Кирххоф Н., Керси Дж. Х.Модель «нокаута» на мышах Mll-AF4 приводит к нарушению регуляции лимфоидной и миелоидной регуляции и гематологическому злокачественному новообразованию. Кровь. 2006. 108: 669–677. [Бесплатная статья PMC] [PubMed] [Google Scholar] 88. Мецлер М., Форстер А., Паннелл Р. и др. Условная модель опухолевого генеза B-клеток MLL-AF4 с использованием инверторной технологии. Онкоген. 2006; 25: 3093–3103. [PubMed] [Google Scholar] 89. Тамай Х., Мияке К., Такатори М. и др. Активированный белок K-Ras ускоряет индуцированную MLL / AF4 лейкемолимфомогенность человека в модели трансгенных мышей.Лейкемия. 2011; 25: 888–891. [PubMed] [Google Scholar] 90. Харрис А. В., Пинкерт Калифорния, Кроуфорд М., Лэнгдон В. Ю., Бринстер Р. Л., Адамс Дж. М.. Трансгенная мышь E. mu-myc. Модель спонтанной лимфомы с высокой заболеваемостью и лейкемии ранних В-клеток. J Exp Med. 1988. 167: 353–371. [Бесплатная статья PMC] [PubMed] [Google Scholar] 91. Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW. Мутации INK4a / ARF ускоряют лимфомагенез и повышают химиорезистентность, отключая p53. Genes Dev. 1999; 13: 2670–2677.[Бесплатная статья PMC] [PubMed] [Google Scholar] 92. Шмитт К.А., Фридман Дж. С., Янг М. и др. Программа старения, контролируемая p53 и p16INK4a, способствует результату лечения рака. Клетка. 2002. 109: 335–346. [PubMed] [Google Scholar] 93. Grabher C, von Boehmer H, Look AT. Активация Notch 1 в молекулярном патогенезе Т-клеточного острого лимфобластного лейкоза. Нат Рев Рак. 2006; 6: 347–359. [PubMed] [Google Scholar] 94. Эллисен Л.В., Берд Дж., Западный округ Колумбия и др. TAN-1, человеческий гомолог гена notch дрозофилы, разрушается хромосомными транслокациями в Т-лимфобластных новообразованиях.Клетка. 1991; 66: 649–661. [PubMed] [Google Scholar] 95. Вен А. П., Феррандо А. А., Ли В. и др. Активирующие мутации NOTCh2 при остром лимфобластном лейкозе Т-клеток человека. Наука. 2004. 306: 269–271. [PubMed] [Google Scholar] 96. Deftos ML, Huang E, Ojala EW, Forbush KA, Bevan MJ. Передача сигналов Notch2 способствует созреванию тимоцитов CD4 и CD8 SP. Иммунитет. 2000. 13: 73–84. [Бесплатная статья PMC] [PubMed] [Google Scholar] 97. Фаулкс Б.Дж., Роби Е.А. Переоценка влияния активированного Notch2 на развитие CD4 и CD8 Т-клеток.J Immunol. 2002; 169: 1817–1821. [PubMed] [Google Scholar] 98. Priceputu E, Bouallaga I, Zhang Y, et al. Структурно различные лиганд-связывающие или лиганд-независимые мутанты Notch2 являются лейкемогенными, но по-разному влияют на развитие тимоцитов, апоптоз и метастазирование. J Immunol. 2006; 177: 2153–2166. [PubMed] [Google Scholar] 99. Berquam-Vrieze KE, Swing DA, Tessarollo L, Dupuy AJ. Характеристика трансгенных мышей, экспрессирующих ассоциированные с раком варианты человеческого NOTCh2. Бытие. 2012; 50: 112–118. [Бесплатная статья PMC] [PubMed] [Google Scholar] 100.Boulos N, Mulder HL, Calabrese CR, et al. Химиотерапевтические агенты предотвращают появление устойчивых к дазатинибу мутаций киназы BCR-ABL на точной мышиной модели острого лимфобластного лейкоза с положительной хромосомой в Филадельфии. Кровь. 2011. 117: 3585–3595. [Бесплатная статья PMC] [PubMed] [Google Scholar] 101. Duy C, Hurtz C, Shojaee S и др. BCL6 позволяет Ph + клеткам острого лимфобластного лейкоза выжить при ингибировании киназы BCR-ABL1. Природа. 2011; 473: 384–388. [Бесплатная статья PMC] [PubMed] [Google Scholar] 102.Ван П.Й., Янг Ф., Чен С.Ю. и др. Биологические свойства лейкозов, возникающих в результате BCR / ABL-опосредованной трансформации, варьируются в зависимости от онтогенетического происхождения и активности гена p19ARF. Кровь. 2008; 112: 4184–4192. [Бесплатная статья PMC] [PubMed] [Google Scholar] 103. Барабе Ф., Кеннеди Дж. А., Хоуп К. Дж., Дик Дж. Э. Моделирование возникновения и прогрессирования острого лейкоза человека у мышей. Наука. 2007; 316: 600–604. [PubMed] [Google Scholar] 104. Вэй Дж., Вундерлих М., Фокс С. и др. Микроокружение определяет судьбу клонов в человеческой модели лейкемии MLL-AF9.Раковая клетка. 2008. 13: 483–495. [Бесплатная статья PMC] [PubMed] [Google Scholar] 105. le Viseur C, Hotfilder M, Bomken S и др. При остром лимфобластном лейкозе у детей бласты на разных стадиях иммунофенотипического созревания обладают свойствами стволовых клеток. Раковая клетка. 2008; 14: 47–58. [Бесплатная статья PMC] [PubMed] [Google Scholar] 106. Чиу П.П., Цзян Х., Дик Дж. Э. Клетки, инициирующие лейкоз при Т-лимфобластном лейкозе человека, проявляют устойчивость к глюкокортикоидам. Кровь. 2010; 116: 5268–5279. [PubMed] [Google Scholar] 107.Отова Б., Сладка М., Панчак А., Маринов И. Биологические характеристики спонтанных трансплантируемых Т-клеточных лимфом у инбредных крыс Sprague-Dawley / детенышей. Transplant Proc. 1997; 29: 1754–1755. [PubMed] [Google Scholar] 108. Отова Б., Вацлавикова Р., Даниелова В. и др. Эффекты паклитаксела, доцетаксела и их комбинаций на подкожные лимфомы у инбредных крыс Sprague-Dawley / Cub. Eur J Pharm Sci. 2006. 29: 442–450. [PubMed] [Google Scholar] 110. Sabaawy HE, Azuma M, Embree LJ, Tsai HJ, Starost MF, Hickstein DD.TELAML1 трансгенная модель рыбок данио предшественника В-клеточного острого лимфобластного лейкоза. Proc Natl Acad Sci U S. A. 2006; 103: 15166–15171. [Бесплатная статья PMC] [PubMed] [Google Scholar] 111. Фрейзер Дж. К., Микер Н. Д., Руднер Л. и др. Модели наследственных Т-клеточных злокачественных новообразований, установленные при фенотипическом скрининге у рыбок данио. Лейкемия. 2009; 23: 1825–1835. [Бесплатная статья PMC] [PubMed] [Google Scholar] 112. Лангенау Д.М., Травер Д., Феррандо А.А. и др. Myc-индуцированный Т-клеточный лейкоз у трансгенных рыбок данио. Наука. 2003. 299: 887–890.[PubMed] [Google Scholar] 113. Feng H, Langenau DM, Madge JA, et al. Индукция теплового шока Т-клеточной лимфомы / лейкемии у условно регулируемых Cre / lox трансгенных рыбок данио. Br J Haematol. 2007. 138: 169–175. [PubMed] [Google Scholar] 114. Feng H, Stachura DL, White RM и др. Клетки Т-лимфобластной лимфомы экспрессируют высокие уровни BCL2, S1P1 и ICAM1, что приводит к блокаде интравазации опухолевых клеток. Раковая клетка. 2010. 18: 353–366. [Бесплатная статья PMC] [PubMed] [Google Scholar] 115. Nowell PC, Hungerford DA.Хромосомные исследования при лейкемии человека. IV. Миелопролиферативный синдром и другие атипичные миелоидные нарушения. J Natl Cancer Inst. 1962; 29: 911–931. [PubMed] [Google Scholar] 116. Goldman JM. Начальное лечение пациентов с ХМЛ. Образовательная программа Hematology Am Soc Hematol. 2009: 453–460. [PubMed] [Google Scholar] 117. Дейли GQ, Ван Эттен Р.А., Балтимор Д. Индукция хронического миелогенного лейкоза у мышей геном P210bcr / abl филадельфийской хромосомы. Наука. 1990; 247: 824–830. [PubMed] [Google Scholar] 118.Ли С., Илария Р.Л., младший, Миллион РП, Дейли Г.К., Ван Эттен Р.А. Формы P190, P210 и P230 онкогена BCR / ABL вызывают аналогичный хронический миелолейкозоподобный синдром у мышей, но обладают различной лимфоидной лейкемогенной активностью. J Exp Med. 1999; 189: 1399–1412. [Бесплатная статья PMC] [PubMed] [Google Scholar] 119. Zhang H, Li H, Xi HS, Li S. HIF1alpha необходим для поддержания выживания стволовых клеток хронического миелоидного лейкоза. Кровь. 2012; 119: 2595–2607. [Бесплатная статья PMC] [PubMed] [Google Scholar] 120. Huettner CS, Koschmieder S, Iwasaki H, et al.Индуцируемая экспрессия BCR / ABL с использованием регуляторных элементов CD34 человека приводит к мегакариоцитарному миелопролиферативному синдрому. Кровь. 2003. 102: 3363–3370. [PubMed] [Google Scholar] 121. Хюттнер К.С., Чжан П., Ван Эттен Р.А., Тенен Д.Г. Обратимость острого В-клеточного лейкоза, вызванного BCR-ABL1. Нат Жене. 2000; 24: 57–60. [PubMed] [Google Scholar] 122. Чу С., Макдональд Т., Лин А. и др. Сохранение стволовых клеток лейкемии у пациентов с хроническим миелолейкозом в длительной ремиссии при лечении иматинибом.Кровь. 2011. 118: 5565–5572. [Бесплатная статья PMC] [PubMed] [Google Scholar] 123. Lozzio BB, Lozzi CB, Machado E. Линия клеток миелогенного (Ph +) лейкоза человека: трансплантация бестимусным мышам. J Natl Cancer Inst. 1976; 56: 627–629. [PubMed] [Google Scholar] 124. Skorski T, Nieborowska-Skorska M, Nicolaides NC и др. Подавление роста лейкозных клеток Philadelphia1 у мышей антисмысловым олигодезоксинуклеотидом BCR-ABL. Proc Natl Acad Sci U S. A. 1994; 91: 4504–4508. [Бесплатная статья PMC] [PubMed] [Google Scholar] 125.Дацци Ф., Хассерджиан Р., Гордон М.Ю. и др. Гемопоэтические клетки ХМЛ нормальной и хронической фаз повторно заселяют костный мозг NOD / SCID с различной кинетикой и представлением клеточных клонов. Hematol J. 2000; 1: 307–315. [PubMed] [Google Scholar] 126. Dohner H, Stilgenbauer S, Benner A и др. Геномные аберрации и выживаемость при хроническом лимфолейкозе. N Engl J Med. 2000; 343: 1910–1916. [PubMed] [Google Scholar] 127. Филлипс Дж. А., Мехта К., Фернандес С., Равече Е. С.. Мышь NZB как модель хронического лимфолейкоза.Cancer Res. 1992; 52: 437–443. [PubMed] [Google Scholar] 128. Равече Э.С., Салерно Э., Скаглионе Б.Дж. и др. Аномальный локус микроРНК-16 с синтенией человеческого 13q14, связанный с ХЛЛ у мышей NZB. Кровь. 2007; 109: 5079–5086. [Бесплатная статья PMC] [PubMed] [Google Scholar] 129. Нардуччи М.Г., Пескармона Э., Лазцери С. и др. Регуляция экспрессии TCL1 в B- и T-клеточных лимфомах и реактивных лимфоидных тканях. Cancer Res. 2000; 60: 2095–2100. [PubMed] [Google Scholar] 130. Бичи Р., Шинтон С.А., Мартин Э.С. и др. Хронический лимфоцитарный лейкоз человека, моделируемый на мышах с помощью направленной экспрессии TCL1.Proc Natl Acad Sci U S. A. 2002; 99: 6955–6960. [Бесплатная статья PMC] [PubMed] [Google Scholar] 131. Агилар-Сантелизес М., Роттенберг М.Э., Левин Н., Меллштедт Н., Йондал М. Экспрессия Bcl-2, Bax и p53 в B-CLL в зависимости от выживаемости и клинического прогрессирования in vitro. Int J Cancer. 1996; 69: 114–119. [PubMed] [Google Scholar] 132. Munzert G, Kirchner D, Stobbe H, et al. Сверхэкспрессия гена фактора некроза опухоли, связанного с рецептором фактора 1, при В-клеточном хроническом лимфоцитарном лейкозе: анализ ингибиторов апоптоза, регулируемых NF-каппа B / Rel.Кровь. 2002; 100: 3749–3756. [PubMed] [Google Scholar] 133. Сапата Дж. М., Краевская М., Морс ХК, 3-й, Цой Й., Рид Дж. Домен TNF-рецепторассоциированного фактора (TRAF) и Bcl-2 взаимодействуют, вызывая малую В-клеточную лимфому / хронический лимфоцитарный лейкоз у трансгенных мышей. Proc Natl Acad Sci U S. A. 2004; 101: 16600–16605. [Бесплатная статья PMC] [PubMed] [Google Scholar] 134. Мохаммад Р.М., Мохамед А.Н., Хамдан М.Ю. и др. Создание модели ксенотрансплантата B-CLL человека: полезность в качестве доклинической терапевтической модели. Лейкемия.1996. 10: 130–137. [PubMed] [Google Scholar] 135. Мохаммад Р.М., Лимварапус С., Хамди Н. и др. Обработка модели ксенотрансплантата de novo устойчивой к флударабину ХЛЛ бриостатином 1 с последующим введением флударабина. Int J Oncol. 1999; 14: 945–950. [PubMed] [Google Scholar] 136. Loisel S, Ster KL, Quintin-Roue I, et al. Создание новой модели человеческого B-CLL-подобного ксенотрансплантата на мышах nude. Leuk Res. 2005; 29: 1347–1352. [PubMed] [Google Scholar] 137. Дуриг Дж., Эбелинг П., Грабеллус Ф. и др. Новая модель ксенотрансплантата, не связанного с ожирением, диабетом и тяжелым иммунодефицитом для хронического лимфолейкоза, отражает важные клинические характеристики заболевания.Cancer Res. 2007; 67: 8653–8661. [PubMed] [Google Scholar] 138. Aydin S, Grabellus F, Eisele L, et al. Изучение роли CD38 и функционально связанных молекулярных факторов риска в модели ксенотрансплантата CLL NOD / SCID. Eur J Haematol. 2011; 87: 10–19. [PubMed] [Google Scholar]Что-нибудь ближе к реальности?
Метастаз рака Ред. Авторская рукопись; доступно в PMC 2014 1 июня.
Опубликован в окончательной отредактированной форме как:
PMCID: PMC3568447
NIHMSID: NIHMS416371
Раздел по гематологии и онкологии, Wake Forest Baptist Health, Уинстон-Сейлем, другие статьи в ЧВК, цитирующих опубликованную статью.
Abstract
Модели на животных сыграли неоценимую роль в усилиях по лучшему пониманию и лечению пациентов, страдающих лейкемией. Несмотря на то, что на основе этих моделей были сделаны важные выводы, следует признать ограничения. В этом обзоре мы выделим различные модели лейкемии на животных и опишем их вклад в улучшение понимания и лечения этих видов рака.
Введение
Лейкемии представляют собой разнообразную группу заболеваний, которые приводят к значительной заболеваемости и смертности.В 2011 году у более 44 600 американцев была диагностирована лейкемия, что привело к 21 780 смертельным исходам [1]. Четыре основных типа составляют 85% всех лейкозов: острый миелоидный лейкоз (AML), острый лимфобластный лейкоз (ALL), хронический миелоидный лейкоз (CML) и хронический лимфолейкоз (CLL). Этим и будет посвящен этот обзор. ХЛЛ является наиболее распространенным заболеванием, но на ОМЛ приходится ~ 42% всех смертей от лейкемии [1]. В результате ОМЛ является наиболее интенсивно изучаемым лейкозом и имеет самую разнообразную группу моделей на животных.Трудно переоценить влияние животных моделей на исследования лейкемии. Начиная с новаторской работы доктора Ллойда Лоу о реакции мышиного лимфоидного лейкоза на антиметаболиты в 1940-х и 1950-х годах до открытия лейкемической стволовой клетки доктором Джоном Диком в 1990-х годах, животные модели были неотъемлемой частью нашего понимания биология лейкозов.
Модели ОМЛ на животных
Введение
За последние 40 лет наше понимание молекулярной патофизиологии ОМЛ значительно выросло (обзор в [2–4]).Заболевание проявляется увеличением незрелых, клональных, миелоидных предшественников, что приводит к прогрессирующей недостаточности костного мозга и, в конечном итоге, к смерти. Несмотря на лучшее понимание генетики ОМЛ, терапия для большинства пациентов осталась неизменной, а общая 5-летняя выживаемость составляет 30-40% [5]. Этот неутешительный показатель выживаемости вдохновил на большую работу по разработке более подходящих моделей на животных для использования при обнаружении новых целей и тестировании новых методов лечения. В результате несколько исследований выявили новые идеи, которые стали возможными только благодаря использованию моделей на животных.Несколько примечательных примеров включают роль взаимодействия клеток AML / стромы костного мозга в устойчивости к химиотерапии [6], способность иммунной системы взаимодействовать с AML и нацеливаться на них [7-8], манипулирование нишей гемопоэтических стволовых клеток посредством AML [9], а также плодотворное наблюдение, что AML существует как иерархия клеток с редкой популяцией лейкемических стволовых клеток [10]. Это наблюдение стало центром теории раковых стволовых клеток, которая теперь распространяется и на многие солидные опухоли (см. Обзор [11]).
Мышиные модели
С 1930-х гг. Мышиные модели наиболее широко использовались для изучения ОМЛ. Первоначальные трансплантируемые модели, индуцированные канцерогенами, уступили место более современным трансгенным, ксенотрансплантатным и мозаичным подходам. В этом разделе будут рассмотрены основные типы мышиных моделей AML.
Канцероген-индуцированные модели
Среди первых описанных моделей мышиного лейкоза были трансплантируемые канцерогенные модели, используемые для тестирования возможных терапевтических агентов (см.).Использование антиметаболитных агентов было впервые протестировано на этих моделях. У них были преимущества, заключающиеся в том, что они могли размножать in vitro и затем вводить их большим когортам реципиентов, у которых впоследствии разовьется лейкемия, что позволило провести испытания многообещающих агентов и графиков. Они были использованы с большим эффектом докторами Скиппером и Шабелем для описания кинетики лейкемогенеза и ответа на терапию, которые остаются основной опорой нашего современного понимания болезни (см. Обзор в [12]).Одной из наиболее широко используемых моделей является линия клеток L1210, выделенная доктором Лоу путем воздействия на мышей DBA / 2 канцерогенного 3-метилхолантрена (3-MC) [13]. Полученный лейкоз был получен от умирающего животного и десятилетиями использовался экспериментально. Дополнительные химически индуцированные линии клеток мышиного лейкоза включают, среди прочего, P388, P1534 и L5178Y (обзор см. В [14]). Однако у этих моделей есть несколько недостатков. Самым сильным из них является тот факт, что патогенез этого лейкоза не имеет отношения к большинству случаев ОМЛ у людей, поскольку у немногих людей лейкоз развивается после воздействия химических агентов.Действительно, по большей части считалось, что некоторые из этих клеточных линий более тесно связаны с лимфоидными злокачественными новообразованиями, чем ОМЛ. Кроме того, генетические основы болезни неизвестны, что ограничивает применимость результатов с использованием этих моделей. Несмотря на эти недостатки, эти модели внесли значительный вклад в лечение ОМЛ, особенно в поддержку использования цитарабина [15], который остается одним из наиболее широко используемых агентов для лечения ОМЛ и ОЛЛ.
Основные методы создания моделей лейкемии на животных.А) Модели, индуцированные канцерогеном. Животные из восприимчивых штаммов подвергаются воздействию канцерогенных химических веществ или ионизирующего излучения. После заражения животные наблюдаются на предмет развития лейкемии. Б) Мозаика, модели транспозонов и вирусов. В мозаичных моделях HSC собирают от мышей-доноров и заражают вирусами, кодирующими интересующий онкоген. Интеграция вируса в геном хозяина приводит к доставке и экспрессии онкогена. В дополнение к доставке онкогенов внедрение вируса может приводить к аберрантной экспрессии генов клеточных протоонкогенов (как показано) или к нарушению клеточного супрессора опухоли, если интеграция происходит внутригенно.Мутагенез транспозонов является результатом аналогичных последствий интеграции транспозонов в геном хозяина. В) Трансгенные модели. Направляющий вектор, который был сконструирован для экспрессии интересующего трансгена, инъецировали или электропорировали в мышиные ES-клетки. ES-клетки, которые интегрировали вектор, затем вводятся в тетраплоидные бластоцисты, которые, в свою очередь, затем вводятся псевдобеременным суррогатным матерям. В качестве альтернативы векторы могут быть непосредственно введены в оплодотворенные зиготы, а затем эти зиготы имплантированы суррогатным матерям.Затем проводят наблюдение за потомством на предмет развития лейкемии. D) Модели ксенотрансплантатов. Первичные образцы пациентов или линии клеток человека можно вводить непосредственно животным-хозяевам с ослабленным иммунитетом. Животные-хозяева часто подвергаются воздействию ионизирующего излучения перед инъекцией образца для подавления остаточной иммунной функции.
Вирусные и транспозонные модели
Одной из самых ранних вирусных моделей ОМЛ является эритробластный ОМЛ, вызываемый вирусами лейкемии Френда, о котором впервые сообщила в 1957 году доктор Шарлотта Френд [16].Она сообщила о присутствии бесклеточного агента, который может серийно передавать эритробластный лейкоз у восприимчивых линий мышей. Позднее было обнаружено, что этот фильтруемый агент представляет собой комбинацию вируса, формирующего очаг селезенки с дефицитом репликации (SFFV), и репликационно-компетентного вируса мышиного лейкоза Friend (MuLV) [17]. SFFV был идентифицирован как причина эритробластного лейкоза [18]; он частично действует путем инсерционного мутагенеза (см.). Интеграция вирусного генома в непосредственной близости от Spi / PU.1 ген приводит к его сверхэкспрессии, управляемой вирусным LTR, что способствует снижению дифференцировки эритробластов, наблюдаемой при заболевании [19]. MuLV был использован для обнаружения новых генов, участвующих в лейкемогенезе AML, в скринингах инсерционного мутагенеза [20]. Дополнительные вирусы лейкемии, индуцирующие AML, были выделены и использованы в скринингах инсерционного мутагенеза с использованием трансгенных моделей, включая вирус MOL4070LTR [21–22]. Вирусно-индуцированный ОМЛ также использовался в доклинической оценке возможных терапевтических средств, в первую очередь на модели спонтанной лейкемии у мышей AKR в результате онкогенного РНК-вируса [23].Помимо вирусно-опосредованного инсерционного мутагенеза, также были разработаны системы на основе транспозонов, в первую очередь система Sleeping Beauty, которая использовалась для идентификации взаимодействующих мутаций [24-25]. Эти модели, индуцированные вирусами и транспозонами, способствовали пониманию лейкемогенеза и идентификации активных терапевтических агентов. Однако их отношение к заболеванию человека сомнительно, поскольку не было получено убедительных доказательств наличия у человека вируса или транспозона, индуцирующего ОМЛ.
Трансгенные модели
ОМЛ характеризуется неслучайными повторяющимися кариотипическими аномалиями, которые, как известно, влияют на прогноз, особенно хромосомные транслокации. Эти транслокации могут быть патогномоничными для определенного подтипа AML, такого как t (15; 17), наблюдаемого при остром промиелоцитарном лейкозе (APL), или могут возникать при различных лейкозах, таких как филадельфийская хромосома, наблюдаемая при CML, ALL и редко AML.
Самые ранние трансгенные модели мышей, созданных с помощью AML, для экспрессии слитых белков, генерируемых этими транслокациями.В трансгенных моделях мышей создаются путем манипуляции с эмбриональными стволовыми (ES) клетками. В классическом подходе ДНК вводится непосредственно в про-ядро оплодотворенных зигот, которые затем вводятся псевдобеременным самкам (см.). Это приводит к нецелевой интеграции трансгена и зависит от экспрессии трансгена для генерации фенотипа. В более современном подходе ДНК электропорируется в ES-клетки, а интегранты отбираются путем экспрессии генов устойчивости к антибиотикам.Выбранные клетки затем вводятся в тетраплоидные бластоцисты, которые, в свою очередь, имплантируются псевдобеременным самкам. Затем потомство подвергают обратному скрещиванию с мышами дикого типа для получения гомозиготно трансгенных мышей. Теперь можно создать векторы, которые нацелены на определенные сайты в геноме посредством гомологичной рекомбинации. Дополнительный уровень сложности может быть добавлен с помощью более новых векторов, которые условно экспрессируют гены в ответ на доксициклин (системы включения / выключения Tet) или рекомбиназу Cre (с использованием систем Lox / Cre).Как только мыши с этими условными аллелями созданы, их можно скрещивать с другими трансгенными животными, которые экспрессируют трансактиватор Tet или рекомбиназу Cre тканеспецифическим образом, чтобы генерировать тканеспецифическую экспрессию трансгена (обзор в [26]). В дополнение к экспрессии онкогенных слитых белков, онкогенные аллели с усилением функции могут быть «сбиты» с их соответствующих нормальных локусов, а опухолевые супрессоры могут быть «выбиты» с использованием тех же подходов.
APL
Ранние модели APL были созданы путем экспрессии гибридного белка, генерируемого t (15; 17), PML-RAR, под контролем различных миелоид-специфичных промоторов [27–29].Эти модели не только воспроизводили гистологический фенотип APL человека, но также ремиссии, наблюдаемые после лечения всей транс-ретиноевой кислотой (ATRA). В трансгенной модели, экспрессирующей слитый белок PLZF-RAR, обнаруженной у пациентов с редким вариантом APL, содержащим t (11; 17), лечение ATRA не вызывало ремиссии у мышей, имитируя клинический сценарий [30]. Несмотря на эти успехи, у модели есть несколько недостатков. Результирующий фенотип зависит от экспрессии встроенного трансгена.При использовании различных миелоид-специфичных промоторов некоторые промоторы давали модели с высокой пенетрантностью и более короткими латентными периодами, в то время как другие выявляли заболевание, более напоминающее миелопролиферативные новообразования, чем острый лейкоз. Действительно, модель, экспрессирующая PML-RARα под контролем промотора CD11b, вообще не развивала лейкоз [27]. Эти модели все еще оставляют вопрос о том, необходимы ли генетические события в дополнение к t (15; 17), чтобы вызывать APL у людей.
AML1-ETO Leukemias
Слитый белок AML1-ETO генерируется t (8; 21), обнаруживаемым преимущественно в AML FAB класса M2.Пациенты с ОМЛ, положительные по транслокации t (8; 21), имеют лучший прогноз и, как правило, поддаются лечению химиотерапевтическими препаратами [31–32]. Слитые гены AML-ETO приводят к гибели эмбрионов при вставке в эндогенный промотор AML1 из-за плохого гематопоэза, аналогично AML1 — / — животных с нокаутом [33]. Это делает стандартные модели AML1-ETO неинформативными. Чтобы обойти это ограничение, доктор Чжан и его коллеги создали индуцибельную трансгенную модель, которая экспрессирует AML1-ETO под контролем репрессора Tet.Несмотря на устойчивую экспрессию трансгена в костном мозге мышей, лейкоз не развился [34]. Впоследствии они обнаружили, что когда мышей, экспрессирующих AML1-ETO (под контролем миелоид-специфического промотора MRP8 ), лечили канцерогеном, N-этил-N-нитрозомочевиной, у 55% животных развился AML [35], в то время как ни один из них не развивался. контрольных мышей. Это открытие согласуется с мнением, что экспрессия AML1-ETO необходима, но недостаточна для лейкемогенеза. В соответствии с этой идеей другие группы обнаружили сотрудничество AML1-ETO с мутациями в тирозинкиназах, таких как TEL-PDGFR, используя мозаичные модели, которые будут обсуждаться ниже [36–37].
MLL Leukemias
Ген смешанной лейкемии (MLL) расположен на хромосоме 11 и часто участвует в сбалансированных транслокациях при остром лейкозе. У него более 40 известных партнеров слияния при остром лейкозе, и он часто ассоциируется с химиорезистентностью и плохим прогнозом [31–32, 38]. Примерно 5% пациентов с острым лейкозом экспрессируют слияния MLL [39–40], хотя это число увеличилось до ~ 70% пациентов, ранее подвергавшихся воздействию антрациклинов и ядов топоизомеразы II.Модели слияния MLL-белков, таких как Mll-lacZ, приводят к AML у одних животных, но не у других. LacZ не имеет известной онкогенной роли, поэтому это предполагает, что одного MLL достаточно, чтобы вызвать онкогенез. В сценарии t (9; 11) AF9 сотрудничает с MLL. Белок AF9 гомологичен продукту ENL из 19p13.3, который является другим геном-партнером для MLL, участвующего в острых лейкозах. Corral et al. использовали гомологичную рекомбинацию в ES-клетках для получения слияния Mll-AF9. Слитый белок, который они создали, зависит от эндогенных контрольных элементов MLL для транскрипции слитого белка.У мышей развился ОМЛ, несмотря на широко распространенную активность промотора MLL [41]. У небольшой группы животных развился ОЛЛ, что согласуется с тем, что наблюдается у людей [42]. Инженерия реципрокной транслокации между хромосомами 9 и 11 для генерации слитого гена MLL-AF9 была создана в одной модели с использованием сайтов LoxP как в локусах MLL, так и в AF9, но у этих мышей не развился лейкоз [43]. Модель продемонстрировала возможность использования систем Lox-Cre для создания транслокаций у мышей.Другая рекуррентная транслокация с участием гена MLL происходит с t (11; 19) и приводит к генерации слитого белка MLL-ENL. Это событие слияния было смоделировано на трансгенных животных с использованием подхода инженерной транслокации [44]. В этом исследовании мышей получали с сайтами LoxP как в генах MLL, так и в генах ENL и скрещивали с мышами, экспрессирующими Cre с гена Lmo2 , специфичного для гематопоэтических клеток. Полученные в результате мыши развили агрессивный, полностью пенетрантный AML, демонстрируя, что подход инженерной транслокации может успешно моделировать AML.
Модели усиления функции, «сбитые с толку»
Несколько онкогенов вовлечены в развитие ОМЛ у людей, в первую очередь тирозинкиназа рецептора Flt3. Мутации Flt3 встречаются у 25% пациентов с ОМЛ и имеют худший прогноз [45]. Наиболее распространенная мутация Flt3 включает в себя создание тандемной дупликации в рамке считывания, так называемая мутация «Flt3 ITD». Flt3 ITD является конститутивно активной формой Flt3, которая вызывает лиганд-независимую передачу сигналов. Была сгенерирована встроенная модель ITD Flt3; у полученных мышей развилось миелопролиферативное новообразование, но не AML; согласуется с другими моделями, которые предполагают, что ITD Flt3 может приводить к усилению пролиферации, но одного недостаточно для развития AML [46].Также согласуется с этим открытием, когда трансгенных мышей, экспрессирующих ITD Flt3, скрещивали с трансгенными животными, экспрессирующими слитый белок NUP98-HOXD13, обнаруженный у пациентов с миелодиспластическим синдромом (МДС) и AML, в результате у полученного потомства развился высокопенетрантный и летальный AML [47].
G-белок Ras является мишенью для активации мутаций при AML. K-ras мутирует примерно в 10–15% AML и в 20–25% N-ras. Была разработана индуцибельная модель K-ras, управляемая его эндогенным промотором [48].В этой модели исследователи создали мутированный аллель K-ras ниже транскрипционной стоп-последовательности, фланкированной сайтами LoxP. Затем эту мышь скрестили с мышью, которая экспрессирует рекомбиназу Cre под контролем интерферон-индуцируемого промотора MX-1 (MX1-Cre). Условная экспрессия K-ras G12D приводит к фатальному миелопролиферативному нарушению, аналогичному таковому в моделях ITD Flt3. Этот вывод согласуется с гипотезой «двух ударов», согласно которой необходимы два изменения, чтобы вызвать ОМЛ [2].Интересно, что когда аналогичная модель была построена с использованием N-ras G12D , был индуцирован гораздо более вялый миелопролиферативный ответ, и мыши умерли от ряда гематологических злокачественных новообразований, демонстрируя, что разные аллели Ras не являются функционально эквивалентными [49].
Наиболее частая мутация при ОМЛ находится в гене неуклофосмина 1 или NPM1 . Он выполняет множество клеточных функций и перемещается между ядерными, ядрышковыми и цитоплазматическими локациями. При AML ~ 35% пациентов имеют мутацию NPM1, которая изменяет клеточную локализацию, что приводит к цитоплазматическому распределению только мутантного белка.Первая модель, которая сбивала мутированную форму NPM1 в эндогенный локус мыши, привела к легкому миелопролиферативному нарушению, но не к AML. Во второй модели наиболее распространенная форма мутации NPM1 была вставлена в локусы мыши, фланкированные сайтами LoxP; Полученные мыши были скрещены с мышами MX1-Cre. После индукции Cre примерно у одной трети двойных трансгенных мышей развился ОМЛ с отсроченным началом. Пенетрантность увеличивалась, а латентность сокращалась с использованием подхода инсерционного мутагенеза на основе транспозонов.Эти модели прояснили роль мутаций, которые приводят к онкогенному усилению функции в развитии AML, и, вероятно, будут продолжать вносить вклад в более глубокое понимание молекулярного патогенеза заболевания.
«Нокаутированные» модели с потерей функции
Некоторые пути роста и выживания могут быть гиперактивированы из-за потери отрицательных регуляторов. Одним из таких примеров при AML является путь киназы PI3, который гиперактивируется у 50–70% пациентов с AML [50].Супрессор опухолей PTEN является негативным регулятором этого пути. Когда были получены трансгенные мыши, которые содержали сайты LoxP, фланкирующие PTEN, и этих животных скрестили с штаммом, специфичным для миелоида Cre, у 11 из 18 потомков развился AML. Полученный AML был моноцитарным по морфологии, а путь киназы PI3 был активирован во многих случаях экстрамедуллярного моноцитарного AML [51]. Негативным регулятором пути Ras является ген NF1 , который стимулирует ГТФазную активность белков Ras, приводя к их инактивации.Потеря NF1 была спроектирована путем конструирования локусов NF1, фланкированных сайтами LoxP, когда этих животных скрещивали с индуцибельной моделью K-Ras G12D , описанной выше. При индуцировании Cre образовывался агрессивный AML с высокой пенетрантностью [52]. Эти модели демонстрируют, что трансгенный подход может применяться в сочетании с системами LoxP-Cre для моделирования потери опухолевых супрессоров при генерации AML.
Мозаичные модели
Как обсуждалось выше, хотя мышиные трансгенные модели внесли большой вклад в наше понимание AML, эти модели имеют существенные недостатки.Количество времени и усилий, необходимых для перехода от конструирования переносчиков к скринингу мышей-основателей и поддержанию размножающейся колонии, может быть непомерно высоким. Альтернативный подход к трансгенной модели состоит в том, чтобы взять гемопоэтические стволовые клетки (HSC) от мышей, манипулировать ими ex vivo и трансплантировать их аутологичным или сингенным реципиентам (см.). Эта модель позволяет быстро генерировать генетически определенные лейкозы, чаще всего за счет ретро- или лентивирусной трансдукции клеток (см. Обзор [26]).
Векторы на основе вируса стволовых клеток мыши (MSCV) являются наиболее часто используемыми ретровирусами с дефицитом репликации при создании моделей мозаичного лейкоза. Эта быстрая и экономичная система широко использовалась при ОМЛ в дополнение к исследованиям, проведенным на трансгенных животных. Например, исследователи ретровирусно трансфицировали и трансдуцировали гибридный белок AML1-ETO в HSC для получения химерных мышей с молекулярными аномалиями, схожими с таковыми у пациентов с этой транслокацией [53].Модели трансплантации с использованием костного мозга, трансдуцированного с помощью AML1-ETO и N-ras G12D , в сингенных реципиентов, показали сотрудничество, ведущее к лейкемии, которая напоминала подтип M2 AML, наиболее часто связанный с AML1-ETO [31]. Мозаичная модель AML, созданная слиянием MLL-ENL и N-ras G12D , напоминала подтип M4-M5, связанный со слияниями MLL. Когда лейкозных мышей лечили цитарабином и антрациклиновой химиотерапией, ответы на две модели значительно различались: мыши MLL-ENL были плохо восприимчивы, в то время как мыши AML-ETO показали высокие показатели ремиссии и даже несколько излечений.Эти ответы отражают клиническую ситуацию и демонстрируют, что эти модели могут резюмировать различные прогностические эффекты этих транслокаций. Кроме того, мозаичная модель использовалась для моделирования прогностических эффектов мутировавших тирозинкиназ. При использовании для изучения ответа на стандартные химиотерапевтические препараты AML, Flt3 ITD ускорял приживление. Эта модель была чувствительна к цитарабину, обеспечивая снижение опухолевой нагрузки и улучшение выживаемости. Доксорубицин не показал такой же эффективности, и было высказано предположение о чистой резистентности при сочетании двух препаратов, как при стандартной индукционной терапии ОМЛ [54].
Модели ксенотрансплантатов
Даже самый клинически агрессивный образец лейкозных клеток от пациента вряд ли вырастет при помещении в культуру, а характеристики клеток могут измениться в зависимости от используемой среды [55–56]. Кроме того, системы культивирования не могут воспроизводить взаимодействия между лейкозными клетками и их стромой и иммунными клетками. Чтобы обойти эти ограничения, были разработаны мышиные модели ксенотрансплантатов, позволяющие приживить первичные образцы пациентов и клеточные линии.
В первых попытках ксенотрансплантации ОМЛ мышам с ослабленным иммунитетом использовались бестимусные голые мыши, несущие мутацию в гене Foxn1 , что привело к почти полному отсутствию функциональных Т-клеток. Ранние эксперименты с использованием первичных образцов пациентов действительно привели к образованию гранулоцитарных сарком, но без вовлечения костного мозга и других органов [57]. В попытке улучшить приживление костного мозга мышей с тяжелым комбинированным иммунодефицитом (SCID) тестировали на их способность приживать образцы пациентов с AML.У этих мышей есть мутации в гене Prkdc и, следовательно, отсутствуют функциональные Т- или В-клетки. Хотя образцы, введенные внутрибрюшинно или имплантированные в капсулы почек, имели значительную скорость отбора, внутривенная инъекция привела к приживлению очень небольшого количества образцов у животных-реципиентов [58]. В важной статье доктор Джон Дик и его коллеги вводили первичные образцы пациентов мышам SCID, которых затем лечили фактором стволовых клеток человека (SCF) и фактором, стимулирующим колонии гранулоцитов-макрофагов (GM-CSF).Это привело к высокому количеству заборов периферической крови или костного мозга пациента, введенных внутривенно; что еще более важно, только часть лейкозных клеток могла успешно прижиться [10]. Это было первое свидетельство иерархии лейкозных клеток, при которой меньшинство населения отвечает за поддержание болезни. С тех пор были выведены мыши с более полным иммунодефицитом. У мышей с диабетом без ожирения (NOD) / SCID нет функциональных B- или T-клеток и снижена активность NK-клеток и моноцитов.У них более высокая скорость приживления первичных образцов пациентов, чем у мышей SCID [59]. Дальнейшее ослабление иммунитета было установлено у мышей NOD / SCID, у которых есть делеции в гене, кодирующем гамма-цепь рецептора интерлейкина 2 (IL2Rγ) (так называемые мыши NSG). Наконец, на этой платформе была создана трансгенная мышь, которая экспрессирует человеческий iL3, GM-CSF и SCF (NSG-tg). Эта мышь значительно улучшила скорость приживления первичных образцов пациентов по сравнению со стандартной мышью NSG [60]. Хотя возможности этих моделей ограничены в отношении взаимодействий лейкозных иммунных клеток, они являются ценными инструментами для оценки доклинической активности многообещающих агентов и взаимодействий лейкозных клеток со стромой.
Немышиные модели
Для изучения ОМЛ использовались другие животные модели, включая крыс. В то время как спонтанные лейкемии у крыс редки, сообщалось о многих моделях, вызванных канцерогенами и радиацией [61–64]. Миеломоноцитарный лейкоз, L5222, был индуцирован в 1967 г., через 326 дней после однократной внутривенной инъекции этилнитрозомочевины (200 мг / кг массы тела) трехмесячной самке крысы BD IX. Этот лейкоз был трансплантирован крысам BD IX и использовался в доклинических исследованиях эффективности [65].Модель APL, созданная на крысах Brown Norway (BNML), использовалась в доклинических исследованиях химиотерапии и трансплантации [66]. Лейкоз был индуцирован у самок крыс линии BN 9,10-диметил-1,2-бензантраценом [67] и имеет характеристики образования колоний, аналогичные характеристикам образования человеческих образцов AML в анализах колоний [68]. Кроме того, эта модель внесла важный вклад в понимание минимальной остаточной болезни при ОМЛ (см. Обзор [69]). Совсем недавно появились сообщения о системах AML, не относящихся к млекопитающим.Одним из примеров является недавний отчет о трансплантации образцов AML человека эмбрионам рыбок данио. В этом исследовании исследователи вводили человеческие клеточные линии AML и образцы пациентов в эмбрионы рыбок данио через 48 часов после оплодотворения. Они использовали от 50 до 200 клеток и увидели снижение лейкемической нагрузки, когда эмбрионы, которым вводили чувствительные клетки, обрабатывали иматинибом [70]. В другом исследовании человеческие APL и CML в клеточных линиях миелоидного бластного кризиса вводили эмбрионам рыбок данио; Затем эмбрионы обрабатывали иматинибом или ATRA, и у них отмечалось снижение лейкемической нагрузки [71].
Другой модельной системой, используемой для изучения AML, является плодовая муха, Drosophila melanogaster . Экспрессия слитого белка AML1-ETO в клетках крови Drosophila нарушала дифференцировку клеток крови, которые полагаются на RUNX1, подобно тому, что наблюдается у пациентов [72]. Эти системы, не относящиеся к млекопитающим, обладают преимуществами более низкой стоимости и повышенной репродуктивной способности в системах с хорошо изученной генетикой. Они могут внести значительный вклад в понимание патогенеза ОМЛ.
Модели ВСЕ на животных
ВСЕ — наиболее распространенная лейкемия у детей, а также встречается у значительного числа взрослых. Для него характерно агрессивное разрастание клональных лимфобластов. ОЛЛ имеет несколько подтипов, включая пре-В-клетки, Т-клетки и зрелые В-клетки или клетки Беркитта. Он может проявляться в основном в виде твердой массы (лимфобластные лимфомы) или с первичным поражением костного мозга (лимфобластные лейкемии). В отличие от AML, сейчас считается, что более 90% детей с диагнозом ALL могут быть излечены [73].Хотя это замечательное достижение является результатом многих направлений исследований, несколько важных открытий в отношении ALL было обнаружено с использованием моделей на животных. Эти выводы включают демонстрацию того, что активация Notch-1 приводит к Т-клеточному ОЛЛ на мышиной модели трансплантата [74], открытие, что у трансгенных мышей, экспрессирующих версию слияния BCR-ABL p190, развился ОЛЛ [75], и доклиническую оценку активность антиметаболитов [76].
Мышиные модели
Как и в случае AML, мышиные модели ALL используют канцероген-индуцированный, вирусно-индуцированный, трансгенный, мозаичный и ксенотрансплантный подходы.Эти модели внесли свой вклад в нынешнее понимание ВСЕГО.
Модели, индуцированные канцерогенами
Как и в случае AML, ранние исследования на мышах основывались на модели, индуцированной канцерогеном. В дополнение к важной клеточной линии L1210, обсужденной выше, существует несколько других канцероген-индуцированных линий ALL, включая клеточную линию L5178Y. Эта клетка является производной DBA / 2-производной метилхолантрен-индуцированной Т-клеточной линии, которая широко использовалась в доклинических исследованиях эффективности и механизмов устойчивости [77–79].
Вирусные модели
Штамм мышей AKR, склонный к лейкемии, был использован для характеристики онкогенных свойств различных вирусов. В результате была получена модель Т-клеточного ОЛЛ у мышей AKR, инфицированных рекомбинантным ретровирусом, нацеленным на тимоциты [80]. Эта модель использовалась для оценки влияния ожирения на прогрессирование ОЛЛ [81]. Кроме того, лимфобластные лейкозы и лимфомы, возникшие в результате мутагенеза вставки ретровируса, позволили охарактеризовать новые гены, участвующие в лимфоидном лейкемогенезе, путем характеристики общих последовательностей вставки вируса [82].Надежность методов была подтверждена, когда было обнаружено, что идентифицированный с помощью скрининга ген Prdm14 сверхэкспрессируется в человеческом B- и T-клеточном ОЛЛ и может инициировать ОЛЛ при сверхэкспрессии вирусной трансдукцией клеток костного мозга мыши [83].
Трансгенные модели
Было опубликовано несколько ВСЕ трансгенных моделей. Эти модели воспроизводят В-клетки, Т-клетки и лейкоз Беркитта / лимфобластные лимфомы.
BCR-ABL
Слитый белок BCR-ABL является результатом транслокации t (9; 22) (q34; q11), также известной как филадельфийская хромосома.Эта транслокация характерна для ХМЛ, но также обнаруживается при В-клеточном ОЛЛ. Существует несколько изоформ этого слитого белка, форма p210 (обнаруживается в основном при CML) и форма p190 (обнаруживается в основном при ALL). Были созданы трансгенные мыши, экспрессирующие изоформу p190 с промотора BCR, но у них была эмбриональная летальность [75]. Напротив, трансгенные мыши, экспрессирующие слияние p190 BCR-ABL с промотора металлотионеина, рождаются живыми и 95% умирают от пре-B-клеточного лейкоза / лимфомы через 35–200 дней [84].Эта модель была использована для тестирования активности новых соединений [85], ингибиторов тирозинкиназы [86] и влияния ожирения на прогрессирование ОЛЛ [81].
MLL Leukemias
Как следует из названия, лейкемия смешанного происхождения, транслокации MLL наблюдаются как при ОМЛ, так и при ОЛЛ. Слитый белок MLL-AF4 наблюдается у пациентов с t (4; 11), и он сильно связан с В-клеточным ОЛЛ. Была создана модель трансгенной мыши, которая экспрессировала MLL-AF4 с эндогенного промотора MLL. Полученные мыши продемонстрировали нерегулируемый лимфоидный и миелоидный рост, а после длительного латентного периода — В-клеточные лимфомы [87].Другая модель была создана с использованием инвертированного аллеля AF4, нацеленного на интрон в гене MLL; при воздействии рекомбиназы Cre происходила инверсия и образование MLL-AF4. Затем мышей скрещивали с различными трансгенными мышами с тканеспецифической экспрессией Cre. Когда присутствовал Cre, у мышей развивались злокачественные новообразования В-клеток с различной латентностью в зависимости от промотора Cre [88]. Трансгенная модель с использованием слитого белка MLL-AF4 человека, экспрессированного из вирусного LTR MSCV, привела к В-клеточным новообразованиям, и латентность могла быть сокращена путем скрещивания этих животных с трансгенными мышами, экспрессирующими K-ras G12D [89].Этот результат подтверждает гипотезу множественного попадания в лейкемогенез.
Eµ-myc
Лимфома / лейкемия Беркитта считается подтипом ОЛЛ и характеризуется транслокациями, в результате которых ген MYC экспрессируется под контролем промоторов тяжелой или легкой цепи иммуноглобулина. В результате болезнь характеризуется высокой скоростью распространения и очень агрессивным поведением. Были получены трансгенные мыши с мышиным геном Myc под контролем промотора тяжелой цепи IgG, имитирующего t (8; 14), наблюдаемый в клинических условиях.В результате у мышей развились агрессивные В-клеточные лимфомы и лейкозы, при этом 90% животных умерли в течение первых 5 месяцев жизни [90]. Эта модель была использована для изучения роли взаимодействующих мутаций в лимфоме / лейкемогенезе [91], а также в основополагающей статье о том, как потеря p53 или избыточная экспрессия BCL2 влияет на химиотерапевтический ответ in vivo [92].
NOTCh2
Рецепторы Notch — это трансмембранные рецепторы, которые при активации лигандом расщепляются на внеклеточные и внутриклеточные части.Внутриклеточная часть (ICN) перемещается в ядро, где она взаимодействует с дополнительными белками, что приводит к транскрипционной активации генов-мишеней (обзор в [93]). NOTCh2 был впервые идентифицирован при транслокации, несущей ALL, Т-лимфоциты человека, t (7; 9) (q34; q34.3). Эта транслокация приводит к образованию гена слияния, в котором промотор TCR управляет экспрессией 3 ’конца локуса NOTCh2 , кодируя только ICN и приводя к конститутивной активации NOTCh2 [94].Эта транслокация обнаруживается только примерно в 1% Т-клеточного ОЛЛ. Однако в последующем сообщении от 50 до 60% образцов Т-клеточного ОЛЛ содержали точечные мутации в NOTCh2, , причастные к патогенезу большинства типов Т-клеточного ОЛЛ [95]. Было создано несколько моделей трансгенных мышей для экспрессии активированных аллелей NOTCh2 из Т-клеточных промоторов [96–98]. У этих мышей изменено развитие тимоцитов CD4, CD8 и развиваются злокачественные опухоли Т-клеток. В более поздней модели используется трансгенная мышь, которая условно экспрессирует две общие точечные мутации в NOTCh2 из своего эндогенного промотора с использованием кассеты Lox-Stop-Lox.Эти мыши продемонстрировали ускоренное развитие злокачественных опухолей Т-клеток в системе мутагенеза транспозонов Спящей красавицы [99].
Mosaic Models
Как и в случае AML, ex-vivo манипуляции с HSC были использованы для создания мышиных моделей ALL. MSCV-векторы, сконструированные для экспрессии слитого белка p190 BCR-ABL, широко использовались для создания моделей ALL B-клеток, положительных по филадельфийской хромосоме. Несколько примеров включают исследование эффектов химиотерапии на развитие мутантов BCR-ABL, устойчивых к ингибиторам тирозинкиназы (TKI) [100], роль BCL6 в устойчивости к TKI [101] и роль супрессора опухолей Arf. в развитии В-клеточного ОЛЛ [102].Онкогены слияния MLL также широко использовались для создания мозаичных ВСЕХ моделей. Исследователи объединили мозаичную модель с моделью ксенотрансплантата и, используя человеческие HSC, полученные из пуповинной крови, инфицировали их вектором, экспрессирующим MLL-ENL на основе MSCV, и вводили их сублетально облученным мышам NOD / SCID. У 26 из 29 мышей, которым вводили инъекцию, развился агрессивный В-клеточный ОЛЛ, демонстрирующий, что MLL-ЭНЛ может индуцировать ОЛЛ в клетках человека [103]. В дополнительном исследовании слияние MLL-AF9 могло продуцировать AML или ALL в CD34 + клетках пуповинной крови человека в зависимости от мыши-реципиента (NSG против NSG-tg) [104].Эти исследования установили возможность создания моделей ОЛЛ из человеческих клеток, размножающихся у мышей с ослабленным иммунитетом, и начали разгадывать, каким образом лейкозы с перегруппировкой MLL могут быть миелоидными или лимфоидными.
Модели ксенотрансплантатов
Те же линии мышей с ослабленным иммунитетом, описанные в разделе ксенотрансплантатов в моделях AML, были использованы для проведения доклинических исследований эффективности во ВСЕХ образцах. Кроме того, они распространили модель лейкозных стволовых клеток на ОЛЛ, наблюдая, что не все В- или Т-клеточные ОЛЛ-клетки могут инициировать лейкемию у мышей NOD / SCID или NSG [105–106].
Немышиные модели
Подразделение инбредных крыс Sprague-Dawley имеет высокую частоту Т-клеточных злокачественных новообразований и использовалось в доклинической оценке терапевтических средств [107–108]. В-клеточный ОЛЛ был получен от инбредных морских свинок и был серийно трансплантирован инбредным реципиентам, но не беспородным животным [109]. Существуют также модели ОЛЛ, не относящиеся к млекопитающим, включая трансгенную модель рыбок данио, основанную на экспрессии слитого белка TELAML1, обнаруженного у пациентов с пре-В-клеточным ОЛЛ с t (12; 21) (p13; q22).Исследователи экспрессировали трансген либо повсеместно (с использованием модифицированного промотора актина), либо ограниченным лимфоидом (с использованием промотора Rag2). Только у рыбок данио, которые экспрессировали трансген повсеместно, развивался ОЛЛ после длительного латентного периода [110]. Скрининг химического мутагенеза с использованием трансгенных рыбок данио, которые экспрессируют усиленный зеленый флуоресцентный белок Т-клеточно-специфическим образом, дал несколько линий, которые имеют наследственный Т-клеточный ОЛЛ [111]. Другая модель ОЛЛ Т-клеток была создана у рыбок данио с использованием мышиного гена c-myc , экспрессируемого под контролем промотора Rag2 рыбок данио.У трансгенных рыб развился Т-клеточный ОЛЛ с короткой латентностью [112]. В элегантной модели доктор Лук и его коллеги использовали трансгенных рыбок данио, которые содержали условный аллель Cre, управляемый промотором теплового шока 70, и скрестили его с трансгенной моделью, содержащей мышиный c-myc , управляемый промотором Rag2 , содержащим промежуточная кассета Lox-stop-DS Red-Lox. Полученное потомство можно было поместить в воду при 37 ° C, чтобы вызвать экспрессию Cre. Иссечение стоп-кассеты может сопровождаться флуоресцентной визуализацией для обнаружения потери DS Red и индукции экспрессии EGFP.У этих животных развилась Т-клеточная лимфобластная лимфома (LBL), экспрессирующая EGFP, которая после лечения тепловым шоком прогрессировала до лейкемии [113]. Эта модель позже была использована для исследования генетических событий, которые приводят к превращению LBL в ALL [114]. У рыбок данио хорошо зарекомендовавшая себя генетика, низкая стоимость и высокие показатели воспроизводства, что делает их идеальной системой для проведения генетических скринингов. Вклад этих моделей только начинается.
Животные модели CML
ХМЛ уникален среди лейкозов тем, что существует единственное онкогенное событие, которое вызывает заболевание — слитый белок BCR-ABL, генерируемый филадельфийской хромосомой.Это была первая известная соматическая хромосомная аномалия, напрямую связанная со злокачественным новообразованием, и стало первым доказательством того, что рак является генетическим заболеванием [115]. Заболевание характеризуется аномальной пролиферацией и накоплением зрелых и созревающих миелоидных клеток. Он имеет три различных клинических фазы: хроническую фазу, ускоренную фазу и фазу взрыва. В прошлом ХМЛ можно было вылечить только трансплантацией аллогенных стволовых клеток, но в последние годы лечение произвело революцию с появлением ингибиторов киназы BCR-ABL, таких как иматиниб, нилотиниб и дазатиниб (обзор в [116]).
Животные модели сыграли уникальную роль в исследованиях ХМЛ. Доктор Ван Эттен показал, что изоформа p210 белка BCR-ABL непосредственно вызывает ХМЛ, используя мозаичную модель мыши. Он использовал ретровирусный вектор, который экспрессировал белок p210 и инфицированные мышиные HSC. Когда инфицированные клетки были трансплантированы сингенным реципиентам, у животных развилось миелопролиферативное заболевание, сильно напоминающее ХМЛ человека [117]. С тех пор эти модели широко использовались для оценки многих аспектов ХМЛ, включая трансформирующий потенциал различных изоформ BCR-ABL [118], генетику прогрессирования заболевания [36] и характеристику стволовых клеток ХМЛ [119]. .
CML также моделировали с использованием трансгенных мышей. В одной модели изоформа p210 экспрессировалась из индуцируемого тетрациклином протомера и скрещивалась с трансгенной мышью, которая экспрессировала tet-трансактиватор с промотора CD34, обеспечивая экспрессию p210 в присутствии доксициклина в компартменте HSC. В результате у мышей развилось миелопролиферативное заболевание с тромбоцитозом [120]. Это противоположно B-клеточному ALL, которое они наблюдали, когда p210 экспрессировался в пре-B-клетках [121], демонстрируя, что фенотип лейкозов, индуцированных BCR-ABL, зависит, по крайней мере, частично от клетки-источника.В дополнение к мозаичным и трансгенным моделям были проведены исследования ксенотрансплантатов с использованием образцов пациентов с ХМЛ. В недавнем примере сохранение популяции стволовых клеток ХМЛ у пациентов в цитогенетической ремиссии было продемонстрировано путем приживления клеток мышам NSG [122]. Это исследование доказывает устойчивость этих лейкозных стволовых клеток даже у пациентов, которые получали терапию TKI в течение многих лет. Первичные образцы пациентов и клеточные линии, полученные от пациентов с ХМЛ в различных фазах, были приживлены бестимусным мышам [123], мышам SCID [124] и мышам NOD / SCID [125].Использование мозаичных, трансгенных и ксенотрансплантных моделей дало неоценимую информацию о молекулярной патологии этого лейкоза.
Животные модели ХЛЛ
ХЛЛ является наиболее распространенной из всех лейкозов и, в отличие от ХМЛ, является генетически гетерогенным заболеванием. Это заболевание пожилых людей, характеризующееся клональной пролиферацией иммунодефицитных CD5 + зрелых В-лимфоцитов. Заболевание считается вялотекущим, при этом средняя выживаемость при низкой стадии заболевания составляет более десяти лет, хотя у подгруппы пациентов будет более скоротечное течение [126].Заболевание может иногда трансформироваться в более агрессивную диффузную крупноклеточную В-клеточную лимфому или, реже, в лимфому Ходжкина посредством процесса, называемого трансформацией Рихтера. К настоящему времени описано несколько моделей ХЛЛ на животных. Как и в случае с другими лейкозами, мыши являются наиболее широко используемой моделью.
Одной из самых ранних моделей мышей с ХЛЛ является спонтанно возникшая модель мышей новозеландских черных (NZB). У мышей NZB развивается клональная пролиферация анеуплоидных лимфоцитов CD5 + B. Заболевание может быть пересажено серийно и иногда превращается в крупноклеточную лимфому, соответствующую трансформации Рихтера, наблюдаемой клинически [127].У этих мышей была обнаружена мутация в локусе микроРНК-16; также наблюдаемый в клетках ХЛЛ человека, считается, что он способствует развитию ХЛЛ [128]. В дополнение к этой спонтанной модели было разработано несколько трансгенных моделей. В трансгенной модели TCL1 человеческий ген TCL1 был клонирован ниже промотора E, что привело к ограничению экспрессии B-клеток на высоком уровне. Ген TCL1 имеет повышенную экспрессию при ряде злокачественных новообразований В-клеток, включая ХЛЛ [129].Полученные мыши развивают клональную или олигоклональную пролиферацию лимфоцитов CD5 + B в более позднем возрасте, имитируя болезнь человека [130]. Дополнительное понимание молекулярной патологии ХЛЛ было получено с помощью двойной трансгенной модели TRAF2DN / Bcl-2. Как Bcl-2, так и факторы, ассоциированные с рецептором фактора некроза опухоли (TRAF), сверхэкспрессируются при злокачественных новообразованиях B-клеток, включая CLL [131–132]. В этой модели исследователи скрестили трансгенных животных, экспрессирующих человеческий BCL2 с промотора тяжелой цепи IgG, с животными, экспрессирующими усеченную форму TRAF2.Полученное потомство развивало прогрессивное накопление клональных CD-экспрессирующих В-клеток, что приводило к сокращению выживаемости [133]. Как и в случае с другими лейкозами, сообщалось о распространении образцов ХЛЛ человека у мышей с ослабленным иммунитетом. Полученная от человека линия клеток CLL, устойчивых к флударабину, WSUCLL, может вызывать опухоли при подкожном введении мышам SCID [134]. Эта модель использовалась для доклинических испытаний [135]. Голые мыши также служили реципиентами клеточных линий CLL человека [136]. Наконец, животных NOD / SCID использовали для приживления первичных образцов пациентов [137].Эта модель была использована для изучения эффектов известных прогностических маркеров ХЛЛ на приживление образца; образцы с неблагоприятными факторами прививаются более агрессивно в этой модели [138]. Первые успехи этой модели делают ее очень привлекательной для будущих доклинических исследований эффективности новых агентов.
Резюме и направления на будущее
Животные модели лейкемии были незаменимы в изучении этих разрушительных болезней. Можно привести убедительный аргумент в пользу того, что наше понимание лейкозов больше, чем любое другое злокачественное новообразование, выиграло от этих модельных систем.Большая часть нынешнего понимания и терапевтических подходов были впервые проверены или открыты с использованием этих моделей. В будущем можно легко представить синергию между различными моделями для дальнейшей разработки новых терапевтических целей. Трансгенные и мозаичные модели имеют очевидную полезность для доклинических испытаний эффективности, а также для изучения взаимодействий стромы и иммунных клеток. У моделей ксенотрансплантатов есть уникальное преимущество, заключающееся в том, что они могут исследовать специфическую биологию клеток человека в in vivo, хотя и в среде с ослабленным иммунитетом.Новые системы, не относящиеся к млекопитающим, могут привести к крупномасштабному генетическому скринингу для выявления критических молекулярных путей, которые могут служить следующим поколением терапевтических мишеней, которые будут оценены на мозаичных, трансгенных и ксенотрансплантных моделях и переведены в клинические испытания для улучшения жизни. наших пациентов с лейкемиями.
Благодарности
Авторы выражают благодарность Карен Кляйн за помощь в редактировании рукописи. Поддержка предоставлена Фондом Дуга Коли по исследованиям лейкемии, Фондом Маккея по исследованиям рака, Фондом исследований Лейта и Фондом Фрэнсис П. Тутуилер.
Библиография
1. Сигел Р., Уорд Э, Броули О., Джемаль А. Статистика рака, 2011: Влияние устранения социально-экономических и расовых различий на преждевременную смерть от рака. CA Cancer J Clin. 2011. 61: 212–236. [PubMed] [Google Scholar] 2. Гиллиланд Д.Г., Джордан Коннектикут, Феликс Калифорния. Молекулярные основы лейкемии. Образовательная программа Hematology Am Soc Hematol. 2004: 80–97. [PubMed] [Google Scholar] 3. Licht JD, Sternberg DW. Молекулярная патология острого миелолейкоза. Образовательная программа Hematology Am Soc Hematol.2005: 137–142. [PubMed] [Google Scholar] 4. Ловенберг Б. Острый миелоидный лейкоз: проблема выявления разнообразия болезней. Образовательная программа Hematology Am Soc Hematol. 2008; 2008: 1–11. [PubMed] [Google Scholar] 5. Dohner H, Estey EH, Amadori S и др. Диагностика и лечение острого миелоидного лейкоза у взрослых: рекомендации международной группы экспертов от имени European Leukemia Net. Кровь. 2010. 115: 453–474. [PubMed] [Google Scholar] 6. Нерви Б., Рамирес П., Реттиг М.П. и др. Хемосенсибилизация острого миелоидного лейкоза (ОМЛ) после мобилизации антагонистом CXCR4 AMD3100.Кровь. 2009. 113: 6206–6214. [Бесплатная статья PMC] [PubMed] [Google Scholar] 7. Jaiswal S, Jamieson CH, Pang WW, et al. CD47 активируется циркулирующими гемопоэтическими стволовыми клетками и лейкозными клетками, чтобы избежать фагоцитоза. Клетка. 2009. 138: 271–285. [Бесплатная статья PMC] [PubMed] [Google Scholar] 8. Маджети Р., Чао М.П., Ализаде А.А. и др. CD47 является неблагоприятным прогностическим фактором и мишенью терапевтических антител для стволовых клеток острого миелоидного лейкоза человека. Клетка. 2009. 138: 286–299. [Бесплатная статья PMC] [PubMed] [Google Scholar] 9.Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Лейкозные клетки создают ниши костного мозга, которые нарушают поведение нормальных кроветворных клеток-предшественников. Наука. 2008; 322: 1861–1865. [PubMed] [Google Scholar] 10. Лапидот Т., Сирард С., Формор Дж. И др. Клетка, инициирующая острый миелоидный лейкоз человека после трансплантации мышам SCID. Природа. 1994; 367: 645–648. [PubMed] [Google Scholar] 11. Джордан CT, Гусман ML, Noble M. Раковые стволовые клетки. N Engl J Med. 2006; 355: 1253–1261. [PubMed] [Google Scholar] 12.Skipper HE, Perry S. Кинетика нормальных и лейкозных популяций лейкоцитов и отношение к химиотерапии. Cancer Res. 1970; 30: 1883–1897. [PubMed] [Google Scholar] 13. Закон LW, Таормина V, Бойл PJ. Ответ острых лимфолейкозов на антагонист пуринов 6-меркаптопурин. Ann N Y Acad Sci. 1954. 60: 244–250. [PubMed] [Google Scholar] 14. Маккормак Э., Брюзруд О., Гьертсен БТ. Животные модели острого миелогенного лейкоза — разработка, применение и перспективы на будущее. Лейкемия. 2005; 19: 687–706.[PubMed] [Google Scholar] 15. Шкипер HE, Schabel FM, Jr, Wilcox WS. Экспериментальная оценка потенциальных противораковых агентов. XXI. Планирование приема арабинозилцитозина для использования его специфичности в S-фазе против лейкозных клеток. Cancer Chemother Rep. 1967; 51: 125–165. [PubMed] [Google Scholar] 16. Френд С. Бесклеточная передача у взрослых швейцарских мышей болезни, имеющей характер лейкемии. J Exp Med. 1957; 105: 307–318. [Бесплатная статья PMC] [PubMed] [Google Scholar] 17. Линемейер Д.Л., Менке Дж. Г., Рускетти С. К., Эванс Л. Х., Сколник Е. М..Последовательности гена оболочки, которые кодируют белок gp52 фокусообразующего вируса селезенки, необходимы для индукции пролиферации эритроидных клеток. J Virol. 1982; 43: 223–233. [Бесплатная статья PMC] [PubMed] [Google Scholar] 18. Вольф Л., Рускетти С. Злокачественная трансформация эритроидных клеток in vivo путем введения нереплицирующегося ретровирусного вектора. Наука. 1985; 228: 1549–1552. [PubMed] [Google Scholar] 19. Back J, Dierich A, Bronn C, Kastner P, Chan S. PU.1 определяет способность эритроидных клеток-предшественников к самообновлению.Кровь. 2004. 103: 3615–3623. [PubMed] [Google Scholar] 20. Erkeland SJ, Valkhof M, Heijmans-Antonissen C, et al. Масштабная идентификация генов болезней, вызывающих острый миелоидный лейкоз. J Virol. 2004; 78: 1971–1980. [Бесплатная статья PMC] [PubMed] [Google Scholar] 21. Кауделл Д., Харпер Д.П., Новак Р.Л. и др. Ретровирусный инсерционный мутагенез идентифицирует активацию Zeb2 как новое лейкемогенное событие взаимодействия у трансгенных мышей CALM-AF10. Кровь. 2010; 115: 1194–1203. [Бесплатная статья PMC] [PubMed] [Google Scholar] 22.Slape C, Hartung H, Lin YW, Bies J, Wolff L, Aplan PD. Ретровирусный инсерционный мутагенез идентифицирует гены, которые взаимодействуют с NUP98-HOXD13 во время лейкемической трансформации. Cancer Res. 2007. 67: 5148–5155. [Бесплатная статья PMC] [PubMed] [Google Scholar] 23. Шкипер HE, Schabel FM, младший, трейдер MW, Laster WR., Младший Ответ на терапию спонтанного, первого прохождения и длинных линий прохождения лейкемии AK. Cancer Chemother Rep., 1969; 53: 345–366. [PubMed] [Google Scholar] 24. Василиу Г.С., Купер Дж. Л., Рад Р. и др.Мутантный нуклеофозмин и взаимодействующие пути приводят к возникновению и прогрессированию лейкемии у мышей. Нат Жене. 2011; 43: 470–475. [Бесплатная статья PMC] [PubMed] [Google Scholar] 25. Коллиер Л.С., Адамс Д.Д., Хакетт С.С. и др. Мутагенез спящей красавицы всего тела может вызвать пенетрантную лейкемию / лимфому и редкую глиому высокой степени злокачественности без связанной с этим эмбриональной летальности. Cancer Res. 2009; 69: 8429–8437. [Бесплатная статья PMC] [PubMed] [Google Scholar] 26. Heyer J, Kwong LN, Lowe SW, Chin L. Генно-инженерные модели мышей, не являющиеся зародышевыми линиями, для трансляционных исследований рака.Нат Рев Рак. 2010; 10: 470–480. [Бесплатная статья PMC] [PubMed] [Google Scholar] 27. Early E, Moore MA, Kakizuka A, et al. Трансгенная экспрессия PML / RARalpha нарушает миелопоэз. Proc Natl Acad Sci U S. A. 1996; 93: 7900–7904. [Бесплатная статья PMC] [PubMed] [Google Scholar] 28. Браун Д., Коган С., Лагассе Е. и др. Трансген PMLRARalpha вызывает острый промиелоцитарный лейкоз у мышей. Proc Natl Acad Sci U S. A. 1997; 94: 2551–2556. [Бесплатная статья PMC] [PubMed] [Google Scholar] 29. Гризолано Дж. Л., Весселшмидт Р. Л., Пеличчи П. Г., Лей Т. Дж..Измененное миелоидное развитие и острый лейкоз у трансгенных мышей, экспрессирующих PML-RAR альфа под контролем регуляторных последовательностей катепсина G. Кровь. 1997. 89: 376–387. [PubMed] [Google Scholar] 30. He LZ, Guidez F, Tribioli C и др. Различное взаимодействие PML-RARalpha и PLZF-RARalpha с корепрессорами определяет дифференциальные ответы на RA в APL. Нат Жене. 1998. 18: 126–135. [PubMed] [Google Scholar] 31. Зубер Дж., Радтке И., Парди Т.С. и др. Мышиные модели ОМЛ человека точно предсказывают ответ на химиотерапию.Genes Dev. 2009; 23: 877–889. [Бесплатная статья PMC] [PubMed] [Google Scholar] 32. Гримуэйд Д., Уокер Х., Оливер Ф. и др. Важность диагностической цитогенетики для исходов при ОМЛ: анализ 1612 пациентов, включенных в исследование MRC AML-10. Рабочие группы Совета медицинских исследований по лейкемии взрослых и детей. Кровь. 1998. 92: 2322–2333. [PubMed] [Google Scholar] 33. Окуда Т., Цай З., Ян С. и др. Экспрессия заблокированного гена лейкемии AML1-ETO ингибирует установление нормального дефинитивного гематопоэза и непосредственно генерирует диспластические гематопоэтические предшественники.Кровь. 1998. 91: 3134–3143. [PubMed] [Google Scholar] 34. Роадс К.Л., Хетерингтон С.Дж., Харакава Н. и др. Анализ роли AML1-ETO в лейкемогенезе с использованием модели индуцибельных трансгенных мышей. Кровь. 2000; 96: 2108–2115. [PubMed] [Google Scholar] 35. Юань Ю., Чжоу Л., Миямото Т. и др. Экспрессия AML1-ETO напрямую участвует в развитии острого миелоидного лейкоза при наличии дополнительных мутаций. Proc Natl Acad Sci U S. A. 2001; 98: 10398–10403. [Бесплатная статья PMC] [PubMed] [Google Scholar] 36.Даш А.Б., Уильямс И.Р., Куток Дж. Л. и др. Мышиная модель бластного кризиса ХМЛ, вызванного сотрудничеством между BCR / ABL и NUP98 / HOXA9. Proc Natl Acad Sci U S. A. 2002; 99: 7622–7627. [Бесплатная статья PMC] [PubMed] [Google Scholar] 37. Гризолано Дж. Л., О’Нил Дж., Каин Дж., Томассон М. Х. Активированная рецепторная тирозинкиназа, TEL / PDGFbetaR, взаимодействует с AML1 / ETO, вызывая острый миелоидный лейкоз у мышей. Proc Natl Acad Sci U S. A. 2003; 100: 9506–9511. [Бесплатная статья PMC] [PubMed] [Google Scholar] 38. Lavau C, Du C, Thirman M, Zeleznik-Le N.Связанные с хроматином свойства CBP, слитого с MLL, вызывают миелодиспластический синдром, который перерастает в миелоидный лейкоз. EMBO J. 2000; 19: 4655–4664. [Бесплатная статья PMC] [PubMed] [Google Scholar] 39. Смотреть на. Онкогенные факторы транскрипции при острых лейкозах человека. Наука. 1997; 278: 1059–1064. [PubMed] [Google Scholar] 40. Daser A, Rabbitts TH. Расширение репертуара гена лейкемии смешанного происхождения MLL в лейкемогенезе. Genes Dev. 2004; 18: 965–974. [PubMed] [Google Scholar] 41. Corral J, Lavenir I, Impey H и др.Ген слияния Mll-AF9, полученный путем гомологичной рекомбинации, вызывает острый лейкоз у химерных мышей: метод создания слитых онкогенов. Клетка. 1996. 85: 853–861. [PubMed] [Google Scholar] 42. Стриссель П.Л., Стрик Р., Томек Р.Дж., Роу Б.А., Роули Д.Д., Железник-Ле, штат Нью-Джерси. Структурные свойства ДНК AF9 аналогичны MLL и могут действовать как горячие точки рекомбинации, приводящие к транслокациям MLL / AF9 и лейкемогенезу. Hum Mol Genet. 2000; 9: 1671–1679. [PubMed] [Google Scholar] 43. Коллинз Э.С., Паннелл Р., Симпсон Э.М., Форстер А., Кролик Т.Х.Межхромосомная рекомбинация генов Mll и Af9, опосредованная cre-loxP в развитии мышей. EMBO Rep. 2000; 1: 127–132. [Бесплатная статья PMC] [PubMed] [Google Scholar] 44. Форстер А., Паннелл Р., Дринан Л. Ф. и др. Разработка de novo реципрокных хромосомных транслокаций, связанных с Mll, для репликации первичных событий рака человека. Раковая клетка. 2003. 3: 449–458. [PubMed] [Google Scholar] 45. Стируолт Д.Л., Радич Ю.П. Роль FLT3 в злокачественных заболеваниях кроветворения. Нат Рев Рак. 2003. 3: 650–665. [PubMed] [Google Scholar] 46.Ли Л., Пилото О., Нгуен Х. Б. и др. Включение внутренней тандемной дупликационной мутации в мышиный FLT3 вызывает миелопролиферативное заболевание на мышиной модели. Кровь. 2008; 111: 3849–3858. [Бесплатная статья PMC] [PubMed] [Google Scholar] 47. Гринблатт С., Ли Л., Слейп С и др. Включение мутации FLT3 / ITD кооперируется со слиянием NUP98-HOXD13 для генерации острого миелоидного лейкоза на модели мышей. Кровь. 2012 [Бесплатная статья PMC] [PubMed] [Google Scholar] 48. Чан ИТ, Гиллиланд Д.Г. Онкогенные K-ras в мышиных моделях миелопролиферативного заболевания и острого миелоидного лейкоза.Клеточный цикл. 2004; 3: 536–537. [PubMed] [Google Scholar] 49. Маккензи К.Л., Дольников А., Миллингтон М., Шоунан Ю., Симондс Г. Мутантные N-расы вызывают миелопролиферативные нарушения и апоптоз у мышей с репопуляцией костного мозга. Кровь. 1999; 93: 2043–2056. [PubMed] [Google Scholar] 50. Мартелли AM, Nyakern M, Tabellini G, et al. Сигнальный путь фосфоинозитид-3-киназы / Akt и его терапевтические последствия для острого миелоидного лейкоза человека. Лейкемия. 2006; 20: 911–928. [PubMed] [Google Scholar] 51. Ю Х, Ли Й, Гао С. и др.Соответствующая мышиная модель моноцитарного лейкоза человека посредством Cre / lox-контролируемой миелоид-специфической делеции PTEN. Лейкемия. 2010; 24: 1077–1080. [Бесплатная статья PMC] [PubMed] [Google Scholar] 52. Каттс Б.А., Шегрен А.К., Андерссон К.М. и др. Дефицит Nf1 взаимодействует с онкогенным K-RAS, вызывая у мышей острый миелоидный лейкоз. Кровь. 2009. 114: 3629–3632. [PubMed] [Google Scholar] 53. de Guzman CG, Warren AJ, Zhang Z, et al. Экспансия гемопоэтических стволовых клеток и отчетливые аномалии развития миелоидов на мышиной модели транслокации AML1-ETO.Mol Cell Biol. 2002; 22: 5506–5517. [Бесплатная статья PMC] [PubMed] [Google Scholar] 54. Парди Т.С., Зубер Дж., Лоу SW. Flt3-ITD изменяет химиотерапевтический ответ in vitro и in vivo p53-зависимым образом. Exp Hematol. 2011; 39: 473–485. e474. [Бесплатная статья PMC] [PubMed] [Google Scholar] 55. Bruserud O, Tore Gjertsen B, Brustugun OT и др. Влияние интерлейкина 10 на бластные клетки, полученные от пациентов с острым миелогенным лейкозом. Лейкемия. 1995; 9: 1910–1920. [PubMed] [Google Scholar] 56. Bruserud O, Gjertsen BT, von Volkman HL.Культура in vitro клеток острого миелогенного лейкоза человека (AML) в бессывороточной среде: исследования нативных бластов AML и клеточных линий AML. J Hematother Stem Cell Res. 2000; 9: 923–932. [PubMed] [Google Scholar] 57. Нара Н., Миямото Т. Прямая и серийная трансплантация острого миелоидного лейкоза человека голым мышам. Br J Рак. 1982; 45: 778–782. [Бесплатная статья PMC] [PubMed] [Google Scholar] 58. Sawyers CL, Gishizky ML, Quan S, Golde DW, Witte ON. Распространение бластных миелоидных лейкозов человека у мышей SCID.Кровь. 1992; 79: 2089–2098. [PubMed] [Google Scholar] 59. Ailles LE, Gerhard B, Kawagoe H, Hogge DE. Характеристики роста предшественников острого миелогенного лейкоза, которые инициируют злокачественное кроветворение у мышей, не страдающих ожирением, диабетом / тяжелым комбинированным иммунодефицитом. Кровь. 1999; 94: 1761–1772. [PubMed] [Google Scholar] 60. Вундерлих М., Чоу Ф.С., Линк К.А. и др. Эффективность ксенотрансплантата AML значительно повышается у мышей NOD / SCID-IL2RG, конститутивно экспрессирующих SCF, GM-CSF и IL-3 человека. Лейкемия. 2010; 24: 1785–1788.[Бесплатная статья PMC] [PubMed] [Google Scholar] 61. Svejda J, Kossey P, Hlavayova E, Svec F. Гистологическая картина лейкемии трансплантируемых крыс, вызванная рентгеновским облучением и метилхолантреном. Новообразования. 1958; 5: 123–131. [PubMed] [Google Scholar] 62. Мориучи Т., Оикава Т., Кодама Т., Ямагути Х., Кобаяси Х. Создание и характеристика трансплантируемого миеломоноцитарного лейкоза крыс. Cancer Res. 1983; 43: 5478–5483. [PubMed] [Google Scholar] 63. Иванкович С., Целлер В.Дж. [Лейкемия L 5222 штамма крыс BD IX.Индуцированная этилнитрозомочевиной моноцитарно-миелоидная трансплантируемая форма для цитохимических и химиотерапевтических исследований] Блют. 1974. 28: 288–292. [PubMed] [Google Scholar] 64. Пирсон Дж. У., Чапарас С. Д., Торгерсен Дж. А., Перк К., Чиригос М. А., Шер Н. А.. Эффект лекарственной терапии против гистологически определенного лейкоза крыс. Cancer Res. 1974; 34: 355–361. [PubMed] [Google Scholar] 65. Zeller WJ, Ivankovic S, Schmahl D. Химиотерапия трансплантируемого острого лейкоза L5222 у крыс. Cancer Res. 1975. 35: 1168–1174. [PubMed] [Google Scholar] 66.Hagenbeek A, Martens AC. Эффективность высоких доз циклофосфамида в сочетании с облучением всего тела при лечении острого миелоцитарного лейкоза: исследования на соответствующей модели крыс. Cancer Res. 1983; 43: 408–412. [PubMed] [Google Scholar] 67. Hagenbeek A, Martens AC. Патогенез крысиной модели острого миелоцитарного лейкоза человека. Haematologica. 1980; 65: 293–308. [PubMed] [Google Scholar] 68. ван Беккум Д.В., ван Остером П., Дике К.А. Образование колоний in vitro при трансплантируемых лейкозах крыс по сравнению с острым миелоидным лейкозом человека.Cancer Res. 1976; 36: 941–946. [PubMed] [Google Scholar] 69. Martens AC, van Bekkum DW, Hagenbeek A. Минимальная остаточная болезнь при лейкемии: исследования на животной модели острого миелоцитарного лейкоза (BNML) Int J Cell Cloning. 1990; 8: 27–38. [PubMed] [Google Scholar] 70. Пруво Б., Жакель А., Дроин Н. и др. Ксенотрансплантат лейкозных клеток в эмбрионе рыбок данио для исследования эффективности лекарств. Haematologica. 2011; 96: 612–616. [Бесплатная статья PMC] [PubMed] [Google Scholar] 71. Коркери Д.П., Деллер Г., Берман Дж. Ксенотрансплантация лейкемии в анализе реакции на химиотерапию рыбок данио in vivo.Br J Haematol. 2011; 153: 786–789. [PubMed] [Google Scholar] 72. Osman D, Gobert V, Ponthan F, Heidenreich O, Haenlin M, Waltzer L. Модель дрозофилы идентифицирует кальпаины как модуляторы человеческого лейкемогенного слитого белка AML1-ETO. Proc Natl Acad Sci U S. A. 2009; 106: 12043–12048. [Бесплатная статья PMC] [PubMed] [Google Scholar] 73. Hunger SP, Lu X, Devidas M и др. Улучшение выживаемости детей и подростков с острым лимфобластным лейкозом в период с 1990 по 2005 год: отчет группы детской онкологии.J Clin Oncol. 2012 [Бесплатная статья PMC] [PubMed] [Google Scholar] 74. Груша В.С., Астер Дж. К., Скотт М.Л. и др. Исключительное развитие Т-клеточных новообразований у мышей с трансплантированным костным мозгом, экспрессирующим активированные аллели Notch. J Exp Med. 1996; 183: 2283–2291. [Бесплатная статья PMC] [PubMed] [Google Scholar] 75. Heisterkamp N, Jenster G, ten Hoeve J, Zovich D, Pattengale PK, Groffen J. Острый лейкоз у трансгенных мышей bcr / abl. Природа. 1990; 344: 251–253. [PubMed] [Google Scholar] 76. Law LW, Dunn TB и др. Наблюдения за действием антагониста фолиевой кислоты на трансплантируемые лимфоидные лейкозы у мышей.J Natl Cancer Inst. 1949; 10: 179–192. [PubMed] [Google Scholar] 77. Тренер DL, Уилок Э.Ф. Характеристика фенотипов клеток L5178Y, выделенных во время прогрессирования состояния покоя опухоли у мышей DBA2. Cancer Res. 1984; 44: 2897–2906. [PubMed] [Google Scholar] 78. Нишимура Т., Муто К., Танака Н. Лекарственная чувствительность устойчивой к адриамицину мутантной сублинии клеток лимфобластомы мыши L5178Y. Дж. Антибиотик (Токио) 1978; 31: 493–495. [PubMed] [Google Scholar] 79. Нисимура Т., Сузуки Х., Муто К., Танака Н. Механизм устойчивости к адриамицину в подлинии клеток лимфобластомы мыши L5178Y.Журнал «Антибиотик» (Токио), 1979; 32: 518–522. [PubMed] [Google Scholar] 80. Клойд М.В., Хартли Дж. В., Роу В.П. Лимфомагенность рекомбинантных вирусов мышиного лейкоза, индуцирующих очаговые клетки норки. J Exp Med. 1980; 151: 542–552. [Бесплатная статья PMC] [PubMed] [Google Scholar] 81. Юн Дж. П., Бехан Дж. В., Хейстеркамп Н. и др. Ожирение, вызванное диетой, ускоряет прогрессирование острого лимфобластного лейкоза в двух моделях на мышах. Cancer Prev Res (Phila) 2010; 3: 1259–1264. [Бесплатная статья PMC] [PubMed] [Google Scholar] 82. Weiser KC, Liu B, Hansen GM и др.Ретровирусные вставки в базе данных VISION идентифицируют молекулярные пути лимфолейкоза и лимфомы мышей. Мамм Геном. 2007. 18: 709–722. [Бесплатная статья PMC] [PubMed] [Google Scholar] 83. Деттман Э.Дж., Симко С.Дж., Аянга Б. и др. Prdm14 инициирует лимфобластный лейкоз после увеличения популяции клеток, напоминающих обычные лимфоидные предшественники. Онкоген. 2011; 30: 2859–2873. [Бесплатная статья PMC] [PubMed] [Google Scholar] 84. Groffen J, Voncken JW, Kaartinen V, Morris C, Heisterkamp N. Ph-позитивный лейкоз: модель трансгенных мышей.Лимфома лейка. 1993; 11 (Приложение 1): 19–24. [PubMed] [Google Scholar] 85. Reichert A, Heisterkamp N, Daley GQ, Groffen J. Лечение Bcr / Abl-положительного острого лимфобластного лейкоза у трансгенных мышей P190 с помощью ингибитора фарнезилтрансферазы SCH66336. Кровь. 2001; 97: 1399–1403. [PubMed] [Google Scholar] 86. Каур П., Фельдхан Н., Чжан Б. и др. Лечение нилотинибом на мышиных моделях лимфобластного лейкоза P190 Bcr / Abl. Молочный рак. 2007; 6: 67. [Бесплатная статья PMC] [PubMed] [Google Scholar] 87. Чен В., Ли К., Хадсон В. А., Кумар А., Кирххоф Н., Керси Дж. Х.Модель «нокаута» на мышах Mll-AF4 приводит к нарушению регуляции лимфоидной и миелоидной регуляции и гематологическому злокачественному новообразованию. Кровь. 2006. 108: 669–677. [Бесплатная статья PMC] [PubMed] [Google Scholar] 88. Мецлер М., Форстер А., Паннелл Р. и др. Условная модель опухолевого генеза B-клеток MLL-AF4 с использованием инверторной технологии. Онкоген. 2006; 25: 3093–3103. [PubMed] [Google Scholar] 89. Тамай Х., Мияке К., Такатори М. и др. Активированный белок K-Ras ускоряет индуцированную MLL / AF4 лейкемолимфомогенность человека в модели трансгенных мышей.Лейкемия. 2011; 25: 888–891. [PubMed] [Google Scholar] 90. Харрис А. В., Пинкерт Калифорния, Кроуфорд М., Лэнгдон В. Ю., Бринстер Р. Л., Адамс Дж. М.. Трансгенная мышь E. mu-myc. Модель спонтанной лимфомы с высокой заболеваемостью и лейкемии ранних В-клеток. J Exp Med. 1988. 167: 353–371. [Бесплатная статья PMC] [PubMed] [Google Scholar] 91. Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW. Мутации INK4a / ARF ускоряют лимфомагенез и повышают химиорезистентность, отключая p53. Genes Dev. 1999; 13: 2670–2677.[Бесплатная статья PMC] [PubMed] [Google Scholar] 92. Шмитт К.А., Фридман Дж. С., Янг М. и др. Программа старения, контролируемая p53 и p16INK4a, способствует результату лечения рака. Клетка. 2002. 109: 335–346. [PubMed] [Google Scholar] 93. Grabher C, von Boehmer H, Look AT. Активация Notch 1 в молекулярном патогенезе Т-клеточного острого лимфобластного лейкоза. Нат Рев Рак. 2006; 6: 347–359. [PubMed] [Google Scholar] 94. Эллисен Л.В., Берд Дж., Западный округ Колумбия и др. TAN-1, человеческий гомолог гена notch дрозофилы, разрушается хромосомными транслокациями в Т-лимфобластных новообразованиях.Клетка. 1991; 66: 649–661. [PubMed] [Google Scholar] 95. Вен А. П., Феррандо А. А., Ли В. и др. Активирующие мутации NOTCh2 при остром лимфобластном лейкозе Т-клеток человека. Наука. 2004. 306: 269–271. [PubMed] [Google Scholar] 96. Deftos ML, Huang E, Ojala EW, Forbush KA, Bevan MJ. Передача сигналов Notch2 способствует созреванию тимоцитов CD4 и CD8 SP. Иммунитет. 2000. 13: 73–84. [Бесплатная статья PMC] [PubMed] [Google Scholar] 97. Фаулкс Б.Дж., Роби Е.А. Переоценка влияния активированного Notch2 на развитие CD4 и CD8 Т-клеток.J Immunol. 2002; 169: 1817–1821. [PubMed] [Google Scholar] 98. Priceputu E, Bouallaga I, Zhang Y, et al. Структурно различные лиганд-связывающие или лиганд-независимые мутанты Notch2 являются лейкемогенными, но по-разному влияют на развитие тимоцитов, апоптоз и метастазирование. J Immunol. 2006; 177: 2153–2166. [PubMed] [Google Scholar] 99. Berquam-Vrieze KE, Swing DA, Tessarollo L, Dupuy AJ. Характеристика трансгенных мышей, экспрессирующих ассоциированные с раком варианты человеческого NOTCh2. Бытие. 2012; 50: 112–118. [Бесплатная статья PMC] [PubMed] [Google Scholar] 100.Boulos N, Mulder HL, Calabrese CR, et al. Химиотерапевтические агенты предотвращают появление устойчивых к дазатинибу мутаций киназы BCR-ABL на точной мышиной модели острого лимфобластного лейкоза с положительной хромосомой в Филадельфии. Кровь. 2011. 117: 3585–3595. [Бесплатная статья PMC] [PubMed] [Google Scholar] 101. Duy C, Hurtz C, Shojaee S и др. BCL6 позволяет Ph + клеткам острого лимфобластного лейкоза выжить при ингибировании киназы BCR-ABL1. Природа. 2011; 473: 384–388. [Бесплатная статья PMC] [PubMed] [Google Scholar] 102.Ван П.Й., Янг Ф., Чен С.Ю. и др. Биологические свойства лейкозов, возникающих в результате BCR / ABL-опосредованной трансформации, варьируются в зависимости от онтогенетического происхождения и активности гена p19ARF. Кровь. 2008; 112: 4184–4192. [Бесплатная статья PMC] [PubMed] [Google Scholar] 103. Барабе Ф., Кеннеди Дж. А., Хоуп К. Дж., Дик Дж. Э. Моделирование возникновения и прогрессирования острого лейкоза человека у мышей. Наука. 2007; 316: 600–604. [PubMed] [Google Scholar] 104. Вэй Дж., Вундерлих М., Фокс С. и др. Микроокружение определяет судьбу клонов в человеческой модели лейкемии MLL-AF9.Раковая клетка. 2008. 13: 483–495. [Бесплатная статья PMC] [PubMed] [Google Scholar] 105. le Viseur C, Hotfilder M, Bomken S и др. При остром лимфобластном лейкозе у детей бласты на разных стадиях иммунофенотипического созревания обладают свойствами стволовых клеток. Раковая клетка. 2008; 14: 47–58. [Бесплатная статья PMC] [PubMed] [Google Scholar] 106. Чиу П.П., Цзян Х., Дик Дж. Э. Клетки, инициирующие лейкоз при Т-лимфобластном лейкозе человека, проявляют устойчивость к глюкокортикоидам. Кровь. 2010; 116: 5268–5279. [PubMed] [Google Scholar] 107.Отова Б., Сладка М., Панчак А., Маринов И. Биологические характеристики спонтанных трансплантируемых Т-клеточных лимфом у инбредных крыс Sprague-Dawley / детенышей. Transplant Proc. 1997; 29: 1754–1755. [PubMed] [Google Scholar] 108. Отова Б., Вацлавикова Р., Даниелова В. и др. Эффекты паклитаксела, доцетаксела и их комбинаций на подкожные лимфомы у инбредных крыс Sprague-Dawley / Cub. Eur J Pharm Sci. 2006. 29: 442–450. [PubMed] [Google Scholar] 110. Sabaawy HE, Azuma M, Embree LJ, Tsai HJ, Starost MF, Hickstein DD.TELAML1 трансгенная модель рыбок данио предшественника В-клеточного острого лимфобластного лейкоза. Proc Natl Acad Sci U S. A. 2006; 103: 15166–15171. [Бесплатная статья PMC] [PubMed] [Google Scholar] 111. Фрейзер Дж. К., Микер Н. Д., Руднер Л. и др. Модели наследственных Т-клеточных злокачественных новообразований, установленные при фенотипическом скрининге у рыбок данио. Лейкемия. 2009; 23: 1825–1835. [Бесплатная статья PMC] [PubMed] [Google Scholar] 112. Лангенау Д.М., Травер Д., Феррандо А.А. и др. Myc-индуцированный Т-клеточный лейкоз у трансгенных рыбок данио. Наука. 2003. 299: 887–890.[PubMed] [Google Scholar] 113. Feng H, Langenau DM, Madge JA, et al. Индукция теплового шока Т-клеточной лимфомы / лейкемии у условно регулируемых Cre / lox трансгенных рыбок данио. Br J Haematol. 2007. 138: 169–175. [PubMed] [Google Scholar] 114. Feng H, Stachura DL, White RM и др. Клетки Т-лимфобластной лимфомы экспрессируют высокие уровни BCL2, S1P1 и ICAM1, что приводит к блокаде интравазации опухолевых клеток. Раковая клетка. 2010. 18: 353–366. [Бесплатная статья PMC] [PubMed] [Google Scholar] 115. Nowell PC, Hungerford DA.Хромосомные исследования при лейкемии человека. IV. Миелопролиферативный синдром и другие атипичные миелоидные нарушения. J Natl Cancer Inst. 1962; 29: 911–931. [PubMed] [Google Scholar] 116. Goldman JM. Начальное лечение пациентов с ХМЛ. Образовательная программа Hematology Am Soc Hematol. 2009: 453–460. [PubMed] [Google Scholar] 117. Дейли GQ, Ван Эттен Р.А., Балтимор Д. Индукция хронического миелогенного лейкоза у мышей геном P210bcr / abl филадельфийской хромосомы. Наука. 1990; 247: 824–830. [PubMed] [Google Scholar] 118.Ли С., Илария Р.Л., младший, Миллион РП, Дейли Г.К., Ван Эттен Р.А. Формы P190, P210 и P230 онкогена BCR / ABL вызывают аналогичный хронический миелолейкозоподобный синдром у мышей, но обладают различной лимфоидной лейкемогенной активностью. J Exp Med. 1999; 189: 1399–1412. [Бесплатная статья PMC] [PubMed] [Google Scholar] 119. Zhang H, Li H, Xi HS, Li S. HIF1alpha необходим для поддержания выживания стволовых клеток хронического миелоидного лейкоза. Кровь. 2012; 119: 2595–2607. [Бесплатная статья PMC] [PubMed] [Google Scholar] 120. Huettner CS, Koschmieder S, Iwasaki H, et al.Индуцируемая экспрессия BCR / ABL с использованием регуляторных элементов CD34 человека приводит к мегакариоцитарному миелопролиферативному синдрому. Кровь. 2003. 102: 3363–3370. [PubMed] [Google Scholar] 121. Хюттнер К.С., Чжан П., Ван Эттен Р.А., Тенен Д.Г. Обратимость острого В-клеточного лейкоза, вызванного BCR-ABL1. Нат Жене. 2000; 24: 57–60. [PubMed] [Google Scholar] 122. Чу С., Макдональд Т., Лин А. и др. Сохранение стволовых клеток лейкемии у пациентов с хроническим миелолейкозом в длительной ремиссии при лечении иматинибом.Кровь. 2011. 118: 5565–5572. [Бесплатная статья PMC] [PubMed] [Google Scholar] 123. Lozzio BB, Lozzi CB, Machado E. Линия клеток миелогенного (Ph +) лейкоза человека: трансплантация бестимусным мышам. J Natl Cancer Inst. 1976; 56: 627–629. [PubMed] [Google Scholar] 124. Skorski T, Nieborowska-Skorska M, Nicolaides NC и др. Подавление роста лейкозных клеток Philadelphia1 у мышей антисмысловым олигодезоксинуклеотидом BCR-ABL. Proc Natl Acad Sci U S. A. 1994; 91: 4504–4508. [Бесплатная статья PMC] [PubMed] [Google Scholar] 125.Дацци Ф., Хассерджиан Р., Гордон М.Ю. и др. Гемопоэтические клетки ХМЛ нормальной и хронической фаз повторно заселяют костный мозг NOD / SCID с различной кинетикой и представлением клеточных клонов. Hematol J. 2000; 1: 307–315. [PubMed] [Google Scholar] 126. Dohner H, Stilgenbauer S, Benner A и др. Геномные аберрации и выживаемость при хроническом лимфолейкозе. N Engl J Med. 2000; 343: 1910–1916. [PubMed] [Google Scholar] 127. Филлипс Дж. А., Мехта К., Фернандес С., Равече Е. С.. Мышь NZB как модель хронического лимфолейкоза.Cancer Res. 1992; 52: 437–443. [PubMed] [Google Scholar] 128. Равече Э.С., Салерно Э., Скаглионе Б.Дж. и др. Аномальный локус микроРНК-16 с синтенией человеческого 13q14, связанный с ХЛЛ у мышей NZB. Кровь. 2007; 109: 5079–5086. [Бесплатная статья PMC] [PubMed] [Google Scholar] 129. Нардуччи М.Г., Пескармона Э., Лазцери С. и др. Регуляция экспрессии TCL1 в B- и T-клеточных лимфомах и реактивных лимфоидных тканях. Cancer Res. 2000; 60: 2095–2100. [PubMed] [Google Scholar] 130. Бичи Р., Шинтон С.А., Мартин Э.С. и др. Хронический лимфоцитарный лейкоз человека, моделируемый на мышах с помощью направленной экспрессии TCL1.Proc Natl Acad Sci U S. A. 2002; 99: 6955–6960. [Бесплатная статья PMC] [PubMed] [Google Scholar] 131. Агилар-Сантелизес М., Роттенберг М.Э., Левин Н., Меллштедт Н., Йондал М. Экспрессия Bcl-2, Bax и p53 в B-CLL в зависимости от выживаемости и клинического прогрессирования in vitro. Int J Cancer. 1996; 69: 114–119. [PubMed] [Google Scholar] 132. Munzert G, Kirchner D, Stobbe H, et al. Сверхэкспрессия гена фактора некроза опухоли, связанного с рецептором фактора 1, при В-клеточном хроническом лимфоцитарном лейкозе: анализ ингибиторов апоптоза, регулируемых NF-каппа B / Rel.Кровь. 2002; 100: 3749–3756. [PubMed] [Google Scholar] 133. Сапата Дж. М., Краевская М., Морс ХК, 3-й, Цой Й., Рид Дж. Домен TNF-рецепторассоциированного фактора (TRAF) и Bcl-2 взаимодействуют, вызывая малую В-клеточную лимфому / хронический лимфоцитарный лейкоз у трансгенных мышей. Proc Natl Acad Sci U S. A. 2004; 101: 16600–16605. [Бесплатная статья PMC] [PubMed] [Google Scholar] 134. Мохаммад Р.М., Мохамед А.Н., Хамдан М.Ю. и др. Создание модели ксенотрансплантата B-CLL человека: полезность в качестве доклинической терапевтической модели. Лейкемия.1996. 10: 130–137. [PubMed] [Google Scholar] 135. Мохаммад Р.М., Лимварапус С., Хамди Н. и др. Обработка модели ксенотрансплантата de novo устойчивой к флударабину ХЛЛ бриостатином 1 с последующим введением флударабина. Int J Oncol. 1999; 14: 945–950. [PubMed] [Google Scholar] 136. Loisel S, Ster KL, Quintin-Roue I, et al. Создание новой модели человеческого B-CLL-подобного ксенотрансплантата на мышах nude. Leuk Res. 2005; 29: 1347–1352. [PubMed] [Google Scholar] 137. Дуриг Дж., Эбелинг П., Грабеллус Ф. и др. Новая модель ксенотрансплантата, не связанного с ожирением, диабетом и тяжелым иммунодефицитом для хронического лимфолейкоза, отражает важные клинические характеристики заболевания.Cancer Res. 2007; 67: 8653–8661. [PubMed] [Google Scholar] 138. Aydin S, Grabellus F, Eisele L, et al. Изучение роли CD38 и функционально связанных молекулярных факторов риска в модели ксенотрансплантата CLL NOD / SCID. Eur J Haematol. 2011; 87: 10–19. [PubMed] [Google Scholar]Лейкоз у животных и человека
Calif Med. 1968 Jan; 108 (1): 14–19.
Эта статья цитируется в других статьях в PMC.Реферат
Рассмотрены общие сравнительные аспекты лейкемии.Лейкоз у взрослого крупного рогатого скота часто встречается в нескольких стадах. У крупного рогатого скота в этих стадах часто наблюдается стойкий лимфоцитоз и повышенное количество атипичных лимфоцитов в крови. Предпринимаются попытки продемонстрировать частоту, с которой это «предлейкозное» или «перилейкемическое» состояние.
С выявлением вирусного возбудителя (ов) у кур, лабораторных грызунов и кошек возрос интерес к лейкемии собак, крупного рогатого скота и других животных, поскольку болезнь этих животных может служить ценными моделями в исследовании и выделение лейкемогенных агентов человека.
Эпидемиологические и клинико-патологические аспекты лейкозов животных имеют сравнительное сходство с самими собой и с лимфоретикулярными новообразованиями человека. Причинный фактор (факторы), вероятно, действует на хозяина, независимо от его вида, аналогичным образом. Маловероятно, но и маловероятно, что лейкоз у домашних животных и лейкоз у человека имеют общие причинно-следственные связи.
Полный текст
Полный текст доступен в виде отсканированной копии оригинальной печатной версии.Получите копию для печати (файл PDF) полной статьи (1,4M) или щелкните изображение страницы ниже, чтобы просмотреть страницу за страницей. Ссылки на PubMed также доступны для Избранные ссылки .
Изображения в этой статье
Щелкните изображение, чтобы увидеть его в увеличенном виде.
Избранные ссылки
Эти ссылки находятся в PubMed. Это может быть не полный список ссылок из этой статьи.
- AHLSTROM CG, JONSSON N, FORSBY N.Саркома Рауса у млекопитающих. Acta Pathol Microbiol Scand Suppl. 1962; Дополнение 154: 127–129. [PubMed] [Google Scholar]
- BELL TM, MASSIE A, ROSS MG, WILLIAMS MC. ВЫДЕЛЕНИЕ РЕОВИРУСА ОТ СЛУЧАЯ ЛИМФОМЫ БЕРКИТТА. Br Med J. 1964, 9 мая; 1 (5392): 1212–1213. [Бесплатная статья PMC] [PubMed] [Google Scholar]
- BIERMAN HR, MARSHALL GJ, WINER ML. ПРЕДИАГНОСТИЧЕСКОЕ СОСТОЯНИЕ ЛИМФОМЫ. ОБНАРУЖЕНИЕ И ОПИСАНИЕ. ДЖАМА. 1963, 19 октября; 186: 185–192. [PubMed] [Google Scholar]
- BURMESTER BR, GROSS MA, WALTER WG, FONTES AK.Патогенность штамма вируса (RPL12), вызывающего висцеральный лимфоматоз птиц и связанные с ним новообразования. II. Взаимосвязи вирусов-хозяев, влияющие на реакцию. J Natl Cancer Inst. 1959 Январь; 22 (1): 103–127. [PubMed] [Google Scholar]
- CROSHAW JE, Jr, ABT DA, MARSHAK RR, HARE WC, SWITZER J, IPSEN J, DUTCHER RM. РОДОСЛОВНЫЕ ИССЛЕДОВАНИЯ ПРИ ЛИМФОСАРКОМЕ КРС. Ann N Y Acad Sci. 1963, 4 ноября; 108: 1193–1202. [PubMed] [Google Scholar]
- EPSTEIN MA, ACHONG BG, BARR YM. ЧАСТИЦЫ ВИРУСА В КУЛЬТУРНЫХ ЛИМФОБЛАСТАХ ЛИМФОМЫ БЕРКИТТА.Ланцет. 1964, 28 марта; 1 (7335): 702–703. [PubMed] [Google Scholar]
- GROSS L. «Спонтанный» лейкоз, развивающийся у мышей C3H после прививки в младенчестве экстрактами AK-лейкемии или AK-эмбрионами. Proc Soc Exp Biol Med. 1951 Янв; 76 (1): 27–32. [PubMed] [Google Scholar]
- GROSS L. Биологические и патогенные свойства вируса лейкемии мышей. Acta Haematol. 1960 May; 23: 259–275. [PubMed] [Google Scholar]
- HARE WC, MCFEELY RA, ABT DA, FEIERMAN JR. ХРОМОСОМНЫЕ ИССЛЕДОВАНИЯ ПРИ ЛИМФОСАРКОМЕ КРС.J Natl Cancer Inst. 1964 июл; 33: 105–118. [PubMed] [Google Scholar]
- ДЖАРРЕТТ В.Ф., МАРТИН В.Б., КРАЙТОН Г.В., ДАЛТОН Р.Г., СТЮАРТ М.Ф. ЭКСПЕРИМЕНТЫ ПЕРЕДАЧИ ЛЕЙКЕМИИ (ЛИМФОСАРКОМА). Природа. 1964, 9 мая; 202: 566–567. [PubMed] [Google Scholar]
- Каваками Т.Г., Тейлен Г.Х., Дангворт Д.Л., Манн Р.Дж., Билл С.Г. Вирусные частицы «С» -типа в плазме крови кошек с лейкемией кошек. Наука. 1967, ноябрь; 158 (3804): 1049–1050. [PubMed] [Google Scholar]
- Moldovanu G, Moore AE, Friedman M, Miller DG.Клеточная передача лимфосаркомы у собак. Природа. 1966, 25 июня; 210 (5043): 1342–1343. [PubMed] [Google Scholar]
- MUNROE JS, WINDLE WF. Опухоли, индуцированные у приматов вирусом саркомы курицы. Наука. 1963, 28 июня; 140 (3574): 1415–1416. [PubMed] [Google Scholar]
- RABOTTI GF, RAINE WA, SELLERS RL. ОПУХОЛИ МОЗГА (ГЛИОМЫ), ВЫЗВАННЫЕ У ХОМЯКОВ ШТАММОМ БРАЯНА ВИРУСА SARCOMA ROUS. Наука. 1965 29 января; 147 (3657): 504–506. [PubMed] [Google Scholar]
- Рикард К. Г., Барр Л. М., Норонья Ф., Догерти Е., 3-е место, сообщение Дж. Э.Частицы вируса С-типа при спонтанном лимфолейкозе у кошек. Cornell Vet. 1967, апрель; 57 (2): 302–307. [PubMed] [Google Scholar]
- СОРЕНСОН Г.Д., ТЕЙЛЕН Г.Х. ЭЛЕКТРОННО-МИКРОСКОПИЧЕСКИЕ НАБЛЮДЕНИЯ ЛИМФОСАРКОМЫ КРС. Ann N Y Acad Sci. 1963, 4 ноября; 108: 1231–1240. [PubMed] [Google Scholar]
- THEILEN GH, SCHALM OW, GILMORE V. Клинические и гематологические исследования лимфосаркомы в стаде крупного рогатого скота. Am J Vet Res. 1961, январь, 22: 23–31. [PubMed] [Google Scholar]
- THEILEN GH, APPLEMAN RD, WIXOM HG.ЭПИЗООТИОЛОГИЯ ЛИМФОСАРКОМЫ КАЛИФОРНИЙСКОГО СКОТА. Ann N Y Acad Sci. 1963, 4 ноября; 108: 1203–1213. [PubMed] [Google Scholar]
- THEILEN GH, DUNGWORTH DL. ЛИМФОСАРКОМА КРС В КАЛИФОРНИИ. 3. ФОРМА ТЕЛЯЧЬИ. Am J Vet Res. 1965 Май; 26: 696–709. [PubMed] [Google Scholar]
- Тейлен Г. Х., Дангворт Д. Л., Харролд Дж. Б., Штрауб О. К.. Исследования передачи лимфосаркомы крупного рогатого скота. Am J Vet Res. 1967 Март; 28 (123): 373–386. [PubMed] [Google Scholar]
- ЗИЛЬБЕР Л.А., КРЮКОВА В.Н. Геморрагическая болезнь крыс, вызванная вирусом саркомы цыплят.Acta Virol. 1957 июль-декабрь; 1 (3-4): 156–160. [PubMed] [Google Scholar]
Здесь представлены статьи из Калифорнийской медицины, любезно предоставленные Издательской группой BMJ
Обновление онкологии: лейкемия собак — Центральная ветеринарная клиника Торонто
Обновление онкологии
Центральная ветеринарная клиника Торонто
Кевин Финора DVM, дипломированный специалист ACVIM (онкология и внутренняя медицина мелких животных)
Лейкоз собак
Лейкемия — это рак иммунной системы и наиболее частая форма рака крови, о которой сообщают у собак.Сообщалось о многих различных типах лейкемии собак. Однако наиболее распространенной формой лейкемии является лимфолейкоз. Лимфоидный лейкоз определяется как аномальное увеличение популяции лимфоцитов в крови. В большинстве случаев местом образования неопластических лимфоцитов является костный мозг, хотя в редких случаях местом производства может быть селезенка. Лейкемия — это, по сути, «лимфома крови». Лимфоидный лейкоз чаще всего отмечается в одной из двух форм; острый лимфобластный лейкоз (ОЛЛ) и хронический лимфолейкоз (ХЛЛ).
ALL — более распространенная форма. ОЛЛ является заболеванием собак молодого и среднего возраста, средний возраст начала заболевания составляет 5,5 лет. ОЛЛ характеризуется внезапным и быстрым образованием крупных лимфобластных клеток. Собаки с ОЛЛ обычно клинически больны, с быстрым появлением клинических признаков. Клинические признаки включают слабость, вялость, анорексию, рвоту, диарею и иногда боль в костях. Во ВСЕХ костный мозг почти исключительно участвует в производстве лимфобластов. В результате может снизиться продукция других клеточных линий, продуцируемых в костном мозге, что может привести к анемии, тромбоцитопении или нейтропении.Развитие цитопении, вторичной по отношению к миелофтизу костного мозга, представляет собой осложнение, которое несет дополнительные риски для пациента. Диагноз лейкемии ставится путем оценки периферической крови, а иногда и проточной цитометрии и / или цитологии костного мозга. Бывают ситуации, когда бывает трудно отличить ОЛЛ от ХЛЛ, и в этих обстоятельствах можно использовать проточную цитометрию для поиска экспрессии CD34. CD34 является маркером ALL, а экспрессия CD34 связана с худшим прогнозом.
ВСЕ трудно поддается лечению. Абсолютно необходима агрессивная терапия. Обычно рекомендуется модифицированный протокол на основе CHOP. Ответ на терапию, как правило, хороший, но непродолжительный. Первоначально количество лимфоцитов будет падать, но редко когда-либо возвращается к нормальному уровню. Очень немногие собаки действительно выживают, чтобы пройти типичный 25-недельный протокол лечения. Прогноз для ALL от серьезного до плохого, а средняя продолжительность жизни превышает 5 месяцев, о которых сообщается редко.
CLL встречается реже по сравнению с ALL.ХЛЛ — это медленно прогрессирующая форма лейкемии. Неопластические лимфоциты хорошо дифференцированы и имеют морфологию, идентичную нормальным лимфоцитам. Неопластические лимфоциты имеют небольшой размер по сравнению с большими бластными клетками, наблюдаемыми при ОЛЛ. ХЛЛ обычно возникает у пожилых собак, средний возраст начала заболевания обычно составляет от 10 до 12 лет. ХЛЛ часто является случайной находкой, обнаруживаемой при рутинном анализе крови, и собаки редко бывают клинически больными. При общем анализе крови повышенное количество лимфоцитов может сопровождаться умеренным снижением других клеточных линий.При патологическом исследовании мазка крови обычно отмечается нормальный вид циркулирующих лимфоцитов. Большинство случаев ХЛЛ имеют Т-клеточное происхождение и медленно прогрессируют. Собаки с ХЛЛ живут намного дольше, чем собаки с ОЛЛ. После постановки диагноза собаки с ХЛЛ должны ежемесячно проверять общий анализ крови. Точка, с которой следует начинать терапию, неясна и спорна. Я отложу начало терапии до тех пор, пока количество лимфоцитов не превысит 60 000 или пока не станут очевидными клинические признаки болезни (несмотря на количество лимфоцитов).Многим собакам может потребоваться год или больше, чтобы их количество увеличилось до уровня, при котором рекомендуется лечение. После начала терапии рекомендуется пероральная терапия преднизоном и хлорамбуцилом. Лечение ХЛЛ носит паллиативный характер, полной ремиссии не ожидается. Количество лимфоцитов упадет и во многих случаях может вернуться к норме. Среднее время выживания для CLL после начала лечения составляет 12 месяцев, 30% собак живут дольше 2 лет.
Д-р Кевин Финора — сертифицированный онколог и терапевт по мелким животным, входящий в медицинскую группу Центральной ветеринарной клиники Торонто.Он доступен для направления и консультаций с понедельника по четверг (включая вечера понедельника и вторника). Пожалуйста, свяжитесь с ним с любыми онкологическими вопросами или проблемами.
Автор: Майкл Гольдштейн, DVM, дипломат ACVIM
Категория: Онкология
Подписаться на новые сообщения:
«предыдущая запись: Подкожный и кожный гемангиосарком у собак // следующая запись: Обновление хирургии: промежностные грыжи»
Лейкоз и другие виды рака крови | Понимание исследований на животных
Лейкемия — это группа раковых заболеваний, поражающих лейкоциты.В здоровом организме белые кровяные тельца борются с инфекцией, но при лейкемии клетки бесконтрольно размножаются в костном мозге. Исследования с использованием мышей сыграли ключевую роль в успешном лечении рака крови.
Лейкемия — это наиболее распространенный вид онкологических заболеваний у детей, на который приходится около трети всех онкологических заболеваний у детей. Еще 25 лет назад диагноз острого лимфолейкоза означал 7 из 10 шансов умереть в течение пяти лет. Теперь, благодаря достижениям в лечении, дети с диагнозом этой формы рака имеют восемь из десяти шансов на выживание.
Важным шагом в поиске эффективных методов лечения любого состояния является понимание того, как оно развивается, и исследования на животных были и продолжают оставаться решающим способом получения этих знаний.
В начале 1970-х исследования на мышах показали, что жизненно важно уничтожить все злокачественные клетки, чтобы избавиться от рака, и что чем раньше можно начать лечение, тем выше шансы на выживание. Этот принцип был руководящим фактором при лечении всех типов рака.
Наше более глубокое понимание генетики помогает создать возможности для лечения многих видов рака, включая лейкоз. В настоящее время проводятся исследования роли генов, вызывающих рак (онкогенов) при лейкемии, с использованием генетически модифицированных (ГМ) мышей. Подобные исследования могут открыть двери для генной терапии лейкемии.
Из доступных в настоящее время методов лечения лейкемии, основным из которых является химиотерапия, животные сыграли важную роль в их развитии.Исследования на животных также помогли разработать новые подходы к лечению лейкемии, такие как иматиниб (гливек), который увеличивает продолжительность жизни пациентов с хроническим миелоидным лейкозом.
Для некоторых, страдающих лейкемией, лучшая надежда — на пересадку костного мозга. Как и при любой форме трансплантации, есть потенциальные проблемы с отторжением. Исследователи работают над способами сделать трансплантаты костного мозга более безопасными и эффективными путем введения иммунной системы человека мышам и изучения того, как она реагирует на привитые клетки.
В то время как показатели выживаемости при лейкемии у детей значительно улучшились, показатели выживаемости взрослых больных лейкемией, к сожалению, не так хороши. Исследования продолжают лучше понимать болезнь и создавать больше и более эффективных методов лечения.
Трансплантаты костного мозга используются для лечения лейкемии, неходжкинской лимфомы и рака крови. Точно так же новые методы лечения стволовыми клетками и моноклональные антитела, такие как ритуксан для лимфомы, стали возможными благодаря исследованиям на мышах.
Для получения дополнительной информации перейдите на сайт AnimalResearch.информационная страница о лейкемии
ИЗОБРАЖЕНИЕ © ISTOCKPHOTO / MYRRHA
Последний раз редактировалось: 18 ноября 2014 17:23
.